Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2015) Volume 2, Issue 4
In the present research work, the thorough experimental and theoretical investigation is made on the crystal compound; Bis (thiourea) Cadmium Bromide (BTCB) by recording FT-IR, FT-Raman and UV Visible spectra. The computational calculations are carried out by HF, CAM-B3LYP, DFT (B3LYP and B3PW91) and LSDA methods with 3-21 G (d, p) basis sets and the corresponding results were tabulated. The compound belongs to orthorhombic crystal class with space group of Pn21a and point group of symmetry C2v. The NLO properties have been studied by calculating average Polarizability and diagonal hyperpolarizability. The physical and chemical properties of the coordination complex due to the Vander Waals link are found to be enriched. The thermodynamical parameters of TGA and DSC are compared with calculated values obtained from NIST thermodynamical program. The variation of specific heat capacity, entropy and enthalpy with respect to different temperature are displayed in the graph and are discussed.
<Keywords: Bis(thiourea) cadmium bromide, BTCB, NLO, Polarizability, Diagonal hyperpolarizability, NIST, TGA-DSC
Now a day, the production of NLO crystal materials using organic compounds with the addition of metal oxides has much attention due to its tremendous electronic and optical applications. Generally, the crystals made up of organic amine derivatives have rich NLO properties. The high symmetry organic amine derivatives; Thiourea has high NLO coefficients with stable physical and chemical properties. Generally, the metal oxide materials are able to have rich semiconducting properties and optical activities. When such metal oxides are coupled with thiourea, the physiochemical, electrical and optical properties are enriched. Due to the symmetrical presence of S and N donors in thiourea, the metal oxides are connected through coordinate covalent bonds strongly. It is a new attempt to fabricate metal organic compound; Bis(thiourea) Cadmium Bromide (BTCB). After careful screening the literature, it is found that no quantum chemical computational work has been made on Bis (thiourea) Cadmium Bromide (BTCB) so far. In this present work, the structural properties, vibrational study, frontier molecular analysis, NMR, UV-Visible spectral investigations have been carried out. The electrical, optical and chemical parameters have been calculated by using HF, CAM, DFT and LSDA method with 3-21 G (d, p) basis set. The optical activity and NLO property analysis have been performed using appropriate quantum computational tools. Other industrial uses of thiourea include the production of flame retardant resins and vulcanization accelerators. Thiourea is used as an auxiliary agent in light-sensitive photocopy paper and almost all other types of copy paper.
• The FT-IR and FT-Raman spectra are recorded in Bruker IFS 66V spectrometer in the range of 4000-100 cm−1 with the spectral resolution is ± 2 cm-1.
• The Thermo Gravimetric Analysis (TGA) and Differential Thermal Analysis (DTA) curves for BTCB are obtained using Simultaneous Thermo gravimetric Analyzer (STA) 409C (NETZSCH) at a heating rate of 10°C/min in nitrogen.
• The absorption spectrum of BTCB is recorded using Varion Cary 5E UV-Vis-NIR spectrophotometer in the range 200-700 nm with high resolution.
In this research work, the most fascinating level of theories RHF and DFT (LSDA, B3LYP and B3PW91) were carried out using the basis sets 3-21G (d, p). All these calculations were performed using GAUSSIAN 09W [1] program. In DFT methods, Becke’s three parameter hybrid function combined with the Lee-Yang-Parr correlation function (B3LYP) [2,3], Becke’s three parameter exact exchange function (B3) [4] combined with gradient-corrected correlation function of Lee, Yang and Parr (LYP) [5,6] and Perdew and Wang (PW91) [6,7] predict the best results for molecular geometry and vibrational frequencies for moderately larger molecules. The calculated frequencies are scaled down to yield the coherent with the observed frequencies. The scaling factors for scaling the harmonic frequencies are 0.864, 0.874, 0.90 and 0.934 for HF/3-21G (d, p) method. For DFT (B3LYP)/3-21G (d, p) basis set, the scaling factors are 0.92, 0.934, 0.96 and 0.98. For DFT (B3PW91)/3-21G (d, p) basis set, the scaling factors are 0.912, 0.92 and 0.93. For CAM (B3LYP)/3-21 G (d, p) basis set, the scaling factors are 0.91, 0.92, 0.93 and 0.96. For LSDA/3-21 G (d, p) level of basis set, the scaling factors such as 0.93, 0.94, 0.96, 0.97 and 0.98 are used. The observed (FT-IR and FT-Raman) and calculated vibrational frequencies with assignments are submitted in Table 1. Experimental and simulated spectra of IR and Raman are displayed in the Figures 1 and 2 respectively.
| Geometrical Parameters |
Methods | ||||
|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | CAM – B3LYP | LSDA | |
| 3-21G | 3-21G | 3-21G | 3-21G | 3-21G | |
| Bond length(Å) | |||||
| (S1-C3) | 1.783 | 1.778 | 1.770 | 1.769 | 1.764 |
| (S1--Cd17) | 2.755 | 2.703 | 2.679 | 2.665 | 2.580 |
| (S2-C4) | 1.782 | 1.778 | 1.770 | 1.769 | 1.764 |
| (S2--Cd17) | 2.755 | 2.703 | 2.679 | 2.665 | 2.580 |
| (C3-N8) | 1.326 | 1.346 | 1.344 | 1.339 | 1.338 |
| (C3-N14) | 1.312 | 1.329 | 1.327 | 1.322 | 1.323 |
| (C4-N5) | 1.312 | 1.329 | 1.327 | 1.322 | 1.323 |
| (C4-N11) | 1.326 | 1.346 | 1.344 | 1.339 | 1.338 |
| (N5-H6) | 1.000 | 1.017 | 1.016 | 1.015 | 1.026 |
| (N5-H7) | 1.008 | 1.035 | 1.037 | 1.033 | 1.061 |
| (N8-H9) | 0.999 | 1.015 | 1.014 | 1.013 | 1.024 |
| (N8-H10) | 0.998 | 1.013 | 1.013 | 1.012 | 1.023 |
| (N11-H12) | 0.999 | 1.015 | 1.014 | 1.013 | 1.024 |
| (N11-H13) | 0.998 | 1.013 | 1.013 | 1.012 | 1.023 |
| (N14-H15) | 1.000 | 1.017 | 1.016 | 1.015 | 1.026 |
| (N14-H16) | 1.008 | 1.035 | 1.037 | 1.033 | 1.061 |
| (Cd17-Br18) | 2.662 | 2.655 | 2.642 | 2.628 | 2.604 |
| (Cd17-Br19) | 2.662 | 2.655 | 2.642 | 2.628 | 2.604 |
| Bond angle(˚) | |||||
| (C3-S1--Cd17) | 110.075 | 106.708 | 106.107 | 106.980 | 96.468 |
| (C4-S2--Cd17) | 110.081 | 106.744 | 106.124 | 106.979 | 96.446 |
| (S1-C3-N8) | 117.151 | 116.711 | 116.682 | 116.737 | 116.481 |
| (S1-C3-N14) | 123.523 | 123.717 | 123.570 | 123.675 | 122.999 |
| (N8-C3-N14) | 119.319 | 119.565 | 119.740 | 119.581 | 120.513 |
| (S2-C4-N5) | 123.525 | 123.711 | 123.570 | 123.678 | 123.001 |
| (S2-C4-N11) | 117.150 | 116.710 | 116.684 | 116.736 | 116.485 |
| (N5-C4-N11) | 119.318 | 119.572 | 119.739 | 119.578 | 120.507 |
| (C4-N5-H6) | 121.774 | 121.48 | 121.650 | 121.517 | 121.490 |
| (C4-N5-H7) | 121.625 | 121.548 | 121.328 | 121.545 | 121.323 |
| (H6-N5-H7) | 116.595 | 116.956 | 117.002 | 116.929 | 117.008 |
| (C3-N8-H9) | 122.596 | 122.604 | 122.665 | 122.593 | 122.652 |
| (C3-N8-H10) | 119.155 | 118.627 | 118.536 | 118.628 | 118.070 |
| (H9-N8-H10) | 118.248 | 118.765 | 118.793 | 118.777 | 119.276 |
| (C4-N11-H12) | 122.597 | 122.608 | 122.667 | 122.594 | 122.647 |
| (C4-N11-H13) | 119.155 | 118.631 | 118.535 | 118.628 | 118.075 |
| (H12-N11-H13) | 118.247 | 118.758 | 118.792 | 118.776 | 119.276 |
| (C3-N14-H15) | 121.775 | 121.476 | 121.652 | 121.515 | 121.486 |
| (C3-N14-H16) | 121.621 | 121.550 | 121.325 | 121.542 | 121.327 |
| (H15-N14-H16) | 116.597 | 116.956 | 117.003 | 116.934 | 117.008 |
| (S1--Cd17--S2) | 96.218 | 98.064 | 98.935 | 98.401 | 111.363 |
| (S1--Cd17-Br18) | 114.660 | 114.380 | 114.439 | 114.556 | 112.572 |
| (S1--Cd17-Br19) | 103.479 | 104.607 | 104.446 | 104.700 | 103.722 |
| (S2--Cd17-Br18) | 103.487 | 104.602 | 104.452 | 104.705 | 103.728 |
| (S2--Cd17-Br19) | 114.664 | 114.474 | 114.434 | 114.549 | 112.571 |
| (Br18-Cd17-Br19) | 121.704 | 118.981 | 118.636 | 118.379 | 113.151 |
| Dihedral angles(˚) | |||||
| (Cd17--S1-C3-N8) | 157.572 | 157.873 | 156.581 | 157.768 | 145.295 |
| (Cd17--S1-C3-N14) | -23.297 | -23.022 | -24.347 | -23.101 | -35.547 |
| (C3-S1--Cd17--S2) | 148.123 | 152.518 | 155.168 | 152.923 | 178.510 |
| (C3-S1--Cd17-Br18) | -103.927 | -97.373 | -94.406 | -96.613 | -65.476 |
| (C3-S1--Cd17-Br19) | 30.878 | 34.527 | 36.944 | 34.697 | 57.193 |
| (Cd17--S2-C4-N5) | -23.266 | -22.940 | -24.300 | -23.081 | -35.541 |
| (Cd17--S2-C4-N11) | 157.604 | 157.961 | 156.623 | 157.783 | 145.299 |
| (C4-S2--Cd17--S1) | 148.059 | 152.297 | 155.120 | 152.924 | 178.547 |
| (C4-S2--Cd17-Br18) | 30.816 | 34.408 | 36.889 | 34.688 | 57.225 |
| (C4-S2--Cd17-Br19) | -103.998 | -97.559 | -94.464 | -96.620 | -65.447 |
| (S1-C3-N8-H9) | 178.030 | 176.100 | 175.740 | 176.662 | 173.723 |
| (S1-C3-N8-H10) | -1.9 | -3.261 | -3.506 | -2.989 | -6.129 |
| (N14-C3-N8-H9) | -1.137 | -3.043 | -3.368 | -2.504 | -5.456 |
| (N14-C3-N8-H10) | 178.931 | 177.594 | 177.384 | 177.843 | 174.691 |
| (S1-C3-N14-H15) | 179.438 | 177.664 | 177.421 | 178.147 | 172.891 |
| (S1-C3-N14-H16) | 0.266 | -0.821 | -0.982 | -0.786 | -2.125 |
| (N8-C3-N14-H15) | -1.449 | -3.255 | -3.534 | -2.746 | -7.983 |
| (N8-C3-N14-H16) | 179.378 | 178.259 | 178.062 | 178.319 | 176.998 |
| (S2-C4-N5-H6) | 179.461 | 177.827 | 177.384 | 178.136 | 172.849 |
| (S2-C4-N5-H7) | 0.288 | -0.721 | -1.000 | -0.803 | -2.154 |
| (N11-C4-N5-H6) | -1.426 | -3.098 | -3.566 | -2.751 | -8.024 |
| (N11-C4-N5-H7) | 179.400 | 178.351 | 178.048 | 178.308 | 176.971 |
| (S2-C4-N11-H12) | 178.050 | 176.238 | 175.734 | 176.642 | 173.661 |
| (S2-C4-N11-H13) | -1.884 | -3.250 | -3.504 | -3.013 | -6.134 |
| (N5-C4-N11-H12) | -1.116 | -2.898 | -3.379 | -2.530 | -5.519 |
| (N5-C4-N11-H13) | 178.948 | 177.611 | 177.381 | 177.813 | 174.684 |
Table 1: Optimized geometrical parameters for Bis (thiourea) Cadmium Bromide(BTCB) computed at HF, DFT (B3LYP& B3PW91), CAM – B3LYP and LSDA with 3-21G (d, p) basis sets.

Figure 1: Crystal view and structure of BTCB.

Figure 2: Experimental [A] and calculated [B, C and D] FT-IR spectra of Bisthiourea Cadmium Bromide (BTCB).
The 1H and 13C NMR isotropic chemical shifts are calculated (Gas, DMSO, Chloroform and CCl4) by the GIAO method with IEFPCM model [8-18] using B3LYP/6-311++G (d, p) level. The electronic properties; HOMO-LUMO energies, absorption wavelengths and oscillator strengths are calculated using B3LYP method of the timedependent DFT (TD-DFT) [19-21], basing on the optimized structure in gas phase and solvent [DMSO, Chloroform and CCl4] mixed phase. Thermodynamic properties have been calculated from 100-1000°C in gas phase using B3LYP/6-311++G (d, p) method. Moreover, the dipole moment, nonlinear optical (NLO) properties, linear polarizabilities and first order hyperpolarizabilities and chemical hardness have also been studied.
Molecular geometry deformational analysis
From the crystal studies, it is observed that, the BTCB belongs to orthorhombic crystal class with space group of Pn21a and point group of symmetry C2v. The compound possesses the symmetrical geometry in which Cd ion is at the tetrahedral coordination site with two Br2 atoms and two NH2 atoms at its top end. This gives rise to a three dimensional bonding network. All thiourea molecules are planar and are equidistant from the central cadmium atom. This structure gives a polymeric character to BTCB molecule with asymmetric units contributing additively to the effective nonlinearity. The molecular structure is optimized by Berny’s optimization algorithm using Gauss view program and is shown in Figure 3. The comparative optimized structural parameters such as bond length, bond angle and dihedral angle are presented in Table 2. The present compound contains cadmium metal atom, bromide atoms and four amino groups.

Figure 3: Experimental [A] and calculated [B, C and D] FT-Raman spectra of Bisthiourea Cadmium Bromide (BTCB).
| S. No. | Symmetry Species C2V |
Observed Frequency(cm-1) | Methods | Vibrational Assignments | |||||
|---|---|---|---|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | CAM – B3LYP | LSDA | |||||
| FT-IR | FT-Raman | 3-21G (d, p) |
3-21G (d, p) |
3-21G (d, p) |
3-21G (d, p) |
3-21G (d, p) |
|||
| 1 | A1 | 3350 s | 3350 s | 3346 | 3349 | 3349 | 3341 | 3348 | (N-H) υ |
| 2 | A1 | 3345 vs | 3345 s | 3345 | 3342 | 3342 | 3337 | 3344 | (N-H) υ |
| 3 | A1 | 3320 vs | 3320 m | 3321 | 3306 | 3321 | 3326 | 3319 | (N-H) υ |
| 4 | A1 | 3300 s | - | 3283 | 3292 | 3296 | 3297 | 3298 | (N-H) υ |
| 5 | A1 | 3295 s | - | 3279 | 3282 | 3293 | 3292 | 3292 | (N-H) υ |
| 6 | A1 | 3290 s | - | 3279 | 3268 | 3257 | 3288 | 3288 | (N-H) υ |
| 7 | A1 | 3250 s | 3250 w | 3247 | 3248 | 3284 | 3249 | 3258 | (N-H) υ |
| 8 | A1 | 3245 vs | 3245 w | 3247 | 3245 | 3229 | 3233 | 3237 | (N-H) υ |
| 9 | A1 | 1610 s | 1610 m | 1622 | 1618 | 1608 | 1609 | 1607 | (N-H) δ |
| 10 | A1 | 1600 m | 1600 m | 1621 | 1594 | 1590 | 1590 | 1588 | (N-H) δ |
| 11 | A1 | 1590 s | - | 1598 | 1577 | 1571 | 1584 | 1586 | (N-H) δ |
| 12 | B2 | 1565 s | - | 1580 | 1553 | 1554 | 1566 | 1569 | (N-H) δ |
| 13 | B2 | 1540 w | 1540 vs | 1533 | 1526 | 1528 | 1526 | 1537 | (N-H) δ |
| 14 | A1 | 1490 m | - | 1476 | 1494 | 1480 | 1477 | 1489 | (N-H) δ |
| 15 | A1 | 1470 s | - | 1453 | 1452 | 1462 | 1453 | 1459 | (N-H) δ |
| 16 | A1 | 1450 w | 1450 s | 1449 | 1448 | 1457 | 1449 | 1453 | (N-H) δ |
| 17 | A1 | 1420 s | - | 1408 | 1409 | 1419 | 1417 | 1413 | (N-H) δ |
| 18 | A1 | 1380 s | - | 1372 | 1376 | 1373 | 1372 | 1380 | (C-N) υ |
| 19 | B2 | 1310 s | 1310 s | 1316 | 1291 | 1326 | 1306 | 1302 | (C-N) υ |
| 20 | B2 | 1260 s | 1260 m | 1256 | 1258 | 1259 | 1251 | 1249 | (C-N) υ |
| 21 | B2 | 1240 m | 1240 m | 1208 | 1211 | 1233 | 1264 | 1239 | (C-S) υ |
| 22 | B2 | 1210 w | - | 1207 | 1209 | 1232 | 1207 | 1208 | (C-S) υ |
| 23 | B2 | 1190 s | - | 1189 | 1187 | 1186 | 1192 | 1185 | (C-N) υ |
| 24 | B2 | 990 m | 990 w | 984 | 995 | 1006 | 983 | 994 | (N-H) γ |
| 25 | B1 | 860 m | - | 859 | 857 | 825 | 858 | 856 | (N-H) γ |
| 26 | B1 | 720 w | 720 w | 735 | 702 | 716 | 718 | 718 | (N-H) γ |
| 27 | A2 | 690 w | - | 709 | 688 | 694 | 684 | 684 | (N-H) γ |
| 28 | A2 | 650 w | - | 661 | 644 | 631 | 658 | 649 | (N-H) γ |
| 29 | A2 | 600 w | - | 602 | 598 | 602 | 594 | 594 | (N-H) γ |
| 30 | A2 | 560 w | - | 562 | 560 | 548 | 557 | 540 | (N-H) γ |
| 31 | B2 | 510 w | - | 534 | 535 | 509 | 513 | 519 | (N-H) γ |
| 32 | B2 | 500 m | - | 498 | 489 | 484 | 479 | 499 | (Cd-Br) υ |
| 33 | A1 | 480 m | 480 w | 495 | 479 | 474 | 457 | 472 | (Cd-Br)υ |
| 34 | A1 | 420 w | - | 431 | 428 | 418 | 415 | 424 | (Cd-S) υ |
| 35 | A1 | 400 m | - | 419 | 403 | 408 | 411 | 402 | (Cd-S) υ |
| 36 | B2 | 390 m | 390 w | 389 | 401 | 405 | 409 | 389 | (C-N) δ |
| 37 | B2 | 380 w | 380 w | 379 | 360 | 365 | 374 | 376 | (C-N) δ |
| 38 | A2 | 360 w | - | 334 | 334 | 343 | 351 | 358 | (C-S) δ |
| 39 | A2 | 310 m | 310 w | 309 | 314 | 326 | 313 | 305 | (C-S) δ |
| 40 | A2 | 290 m | 290 w | 304 | 290 | 295 | 288 | 302 | (Cd-Br) δ |
| 41 | B2 | 270 w | - | 266 | 244 | 248 | 255 | 262 | (Cd-Br) δ |
| 42 | B2 | 220 w | 220 vw | 229 | 241 | 246 | 236 | 228 | (Cd-S) δ |
| 43 | B1 | 200 w | 200 vw | 194 | 196 | 198 | 202 | 206 | (Cd-S) δ |
| 44 | B1 | 170 w | - | 140 | 148 | 152 | 152 | 176 | (C-N) γ |
| 45 | B1 | 165 w | - | 138 | 140 | 142 | 142 | 158 | (C-N) γ |
| 46 | B1 | 140 w | - | 132 | 132 | 130 | 136 | 138 | (C-S) γ |
| 47 | B1 | 130 w | - | 126 | 124 | 124 | 130 | 128 | (C-S) γ |
| 48 | B1 | 120 w | 120 vw | 102 | 106 | 106 | 108 | 112 | (Cd-Br) γ |
| 49 | B1 | 110 w | 110 vw | 80 | 82 | 80 | 84 | 82 | (Cd-Br) γ |
| 50 | B1 | 100 w | - | 28 | 32 | 32 | 28 | 38 | (Cd-S) γ |
| 51 | B1 | 90 w | - | 18 | 24 | 24 | 22 | 26 | (Cd-S) γ |
s – Strong; m- Medium; w – weak; as- Asymmetric; s – symmetric; υ – stretching;
α –deformation, δ - In plane bending; γ-out plane bending; τ – Twisting:
Table 2: Observed and calculated vibrational frequencies of Bis (thiourea) Cadmium Bromide (BTCB) with HF, DFT (B3LYP& B3PW91), CAM – B3LYP and LSDA with 3-21G (d, p) basis sets.
The zero point vibrational energy of the compound in different level of calculations such as HF/LSDA/B3LYP/B3PW91/CAM-B3LYP with 3-21 G(d, p) basis set is 86.85, 78.35, 80.02, 80.55 and 81.43 Kcal/ Mol, respectively. The calculated energy of HF is greater than DFT method since the assumed ground state energy in HF is greater than the true energy. Since the existence of coordinate covalent bond between organic element and metal, molecular structure belongs to multiple planes with respect to Cadmium bromide. The thiourea on both sides are somewhat tilted in order to sustain their equal distribution of charges of Br and H. Since the present compound is composed of metal atom and organic complex, the entire atoms are connected by covalent and co-ordination covalent bonds. Particularly, cadmium metal atom is connected with couple of thiourea by Vander walls bonds. The metal ions acted as a bridge for both thiourea. Normally, the metal ions make dative bond with organic atoms to form organo-metallic compound due to which the considerable amount of energy is released and make a crystal very strong.
In the experimental method, the bond lengths of C-S and C-N vibrations are 1.720 and 1.340 Å whereas in the calculation method, the corresponding bond lengths are 1.778 and 1.346 Å respectively. The internuclear distance of N5-H6=N14-H15 is 0.018 Å and this value is greater than that of N5-H7=N14-H16. This variation exists due to the attraction of Br and H atoms. Moreover, in this compound, both the amino groups are coupled with carbon atom in symmetrical manner. However, the bond distance of C-N is differed by 0.017 Å. This difference occurs between them is due to the attraction of H by Br. There exists a double bond between C and S atoms usually, but only one bond exists due to 2 lone pair of electrons which are transferred from the ligand (S) to the metal (Cd). The calculated bond length of Cd-S is 2.703 Å and this peculiar bond is called coordination covalent bond which has very high bond length when compared with others. It is concluded that from these calculated parameters, this organo-metallic compound is very strong due to the existence of complex bonds.
The coordination covalent bond is an anisotropic bond which is altered at any instant due to the relative orientation of the molecules. The induction and dispersion interactions are always attractive, irrespective of the orientation of the molecules, but the electrostatic interaction between the metal and organic atoms changes sign with respect to the charges of the atom. Thus, the electrostatic force of attraction depending on the charges of the molecule has restricted the bond length which is existed between the metal and organic atoms. Such a force of attraction between metal and organic atom also affect the surrounding atoms of opposite signs. The highly electronegative bromine atoms are attracted much more by the highly positive cadmium atoms.
Mulliken charge analysis: The Mulliken charge is used to understand the charge distribution on the chemical bonding because it facilitates positive and negative regions in the molecular space, at which the protons and electrons concentrate. Thus chemically significant regions; bonds can be identified; also gives the narration of the mechanism of electrophilic and nucleophilic substitutions. Normally, the charges are distributed evenly over the molecule which leads to be neutral. Whenever the substitutions are added to the molecule, the charge distribution is completely altered with respect to the substitution. Here, the negative charges are accumulated over the N atoms in thiourea even after the cadmium bromide is added. When the highly electronegative bromine atoms are coupled with positive cadmium atom, the high degree of Br-Cd-Br dipoles are formed. The remaining C and H of the molecule have positive space. Since the addition of Cd-Br have been occurred in thiourea, the sulphur atoms become low order negative that is almost neutral. The Mulliken charges of each atom are presented in Table 3. Thus the entire charge levels of the molecule are altered on par with due to the substitution. Simultaneously, the chemical property has also changed for the same. The Mulliken charge analysis diagram is displayed in the Figure 4.

Figure 4: Mulliken charge of Bisthiourea Cadmium Bromide.
| Atom | Mullikan Charges |
|---|---|
| S1 | - 0.124 |
| S2 | - 0.124 |
| C3 | 0.475 |
| C4 | 0.475 |
| N5 | - 0.911 |
| N8 | - 0.891 |
| N11 | - 0.891 |
| N14 | - 0.911 |
| H6 | 0.369 |
| H7 | 0.434 |
| H9 | 0.369 |
| H10 | 0.410 |
| H12 | 0.369 |
| H13 | 0.410 |
| H15 | 0.369 |
| H16 | 0.434 |
| Cd17 | 0.781 |
| Br18 | - 0.522 |
| Br19 | - 0.522 |
Table 3: Mulliken charges HF / 3-21G (d, p) level for Bis (thiourea) Cadmium Bromide (BTCB).
Vibrational assignments
The BTCB molecule has 19 atoms and 51 normal vibrational modes. The molecule possesses C2V point group symmetry, which shows that all vibrational modes of BTCB molecule are both infrared (IR) and Raman active. In HF and DFT calculations, the calculated vibrational frequencies were scaled by scaling factors. The different calculated vibrational modes were scaled by different scaling factors for better agreement with the experimental values. On the basis of C2V symmetry, the 51 fundamental vibrations of the molecule can be distributed as
Γ Vib=19A1+7A2+11B1+14B2
A1 and B2 irreducible representations correspond to stretching, ring deformation and in plane bending vibrations while A2 and B1 correspond to ring, torsion and out of plane bending vibrations. The harmonic vibrational frequencies (unscaled and scaled) are calculated at B3LYP and B3PW91 levels using the triple split valence basis set along with the diffuse and polarization functions; 3-21 G (d, p). The observed FT-IR and FT-Raman frequencies for various modes of vibrations have been presented in Tables 2 and 4 respectively. In this, the calculated frequencies are compared with the experimental values and this work reveals the over estimation of the calculated vibrational modes due to the neglect of a harmonicity and change of state of real system Table 5. Also, these computational calculations are carried out for frequency analysis to get the spectroscopic indication of the BTCB.
| S. No | Observed frequency | Calculated frequency | ||||
|---|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | CAM - B3LYP | LSDA | ||
| 3-21G | 3-21G | 3-21G | 3-21G | 3-21G | ||
| 1 | 3350 | 3873 | 3641 | 3669 | 3668 | 3562 |
| 2 | 3345 | 3872 | 3641 | 3669 | 3668 | 3562 |
| 3 | 3320 | 3800 | 3540 | 3572 | 3577 | 3458 |
| 4 | 3300 | 3800 | 3540 | 3572 | 3577 | 3458 |
| 5 | 3295 | 3752 | 3514 | 3541 | 3548 | 3433 |
| 6 | 3290 | 3752 | 3514 | 3541 | 3548 | 3433 |
| 7 | 3250 | 3608 | 3185 | 3166 | 3233 | 2884 |
| 8 | 3245 | 3608 | 3185 | 3166 | 3233 | 2891 |
| 9 | 1610 | 1878 | 1734 | 1730 | 1749 | 1657 |
| 10 | 1600 | 1877 | 1733 | 1729 | 1748 | 1655 |
| 11 | 1590 | 1829 | 1690 | 1690 | 1704 | 1636 |
| 12 | 1565 | 1829 | 1689 | 1690 | 1703 | 1635 |
| 13 | 1540 | 1642 | 1558 | 1576 | 1590 | 1569 |
| 14 | 1490 | 1641 | 1557 | 1575 | 1589 | 1568 |
| 15 | 1470 | 1556 | 1424 | 1434 | 1453 | 1403 |
| 16 | 1450 | 1552 | 1420 | 1429 | 1449 | 1398 |
| 17 | 1420 | 1204 | 1110 | 1118 | 1134 | 1104 |
| 18 | 1380 | 1204 | 1110 | 1117 | 1134 | 1104 |
| 19 | 1310 | 1197 | 1104 | 1105 | 1117 | 1068 |
| 20 | 1260 | 1197 | 1104 | 1105 | 1117 | 1068 |
| 21 | 1240 | 967 | 865 | 881 | 903 | 905 |
| 22 | 1210 | 966 | 864 | 880 | 901 | 902 |
| 23 | 1190 | 820 | 711 | 719 | 745 | 710 |
| 24 | 990 | 820 | 711 | 719 | 745 | 710 |
| 25 | 860 | 735 | 675 | 689 | 692 | 685 |
| 26 | 720 | 735 | 675 | 688 | 691 | 684 |
| 27 | 690 | 709 | 620 | 631 | 658 | 622 |
| 28 | 650 | 708 | 620 | 631 | 658 | 622 |
| 29 | 600 | 602 | 539 | 548 | 556 | 540 |
| 30 | 560 | 602 | 539 | 548 | 557 | 540 |
| 31 | 510 | 534 | 471 | 476 | 480 | 472 |
| 32 | 500 | 534 | 470 | 475 | 479 | 471 |
| 33 | 480 | 495 | 428 | 431 | 457 | 425 |
| 34 | 420 | 494 | 428 | 431 | 457 | 424 |
| 35 | 400 | 419 | 403 | 408 | 411 | 402 |
| 36 | 390 | 417 | 401 | 405 | 409 | 402 |
| 37 | 380 | 218 | 212 | 215 | 220 | 235 |
| 38 | 360 | 192 | 197 | 202 | 207 | 224 |
| 39 | 310 | 178 | 185 | 192 | 196 | 218 |
| 40 | 290 | 175 | 171 | 174 | 180 | 189 |
| 41 | 270 | 133 | 144 | 146 | 150 | 164 |
| 42 | 220 | 132 | 142 | 145 | 148 | 163 |
| 43 | 200 | 97 | 98 | 99 | 101 | 103 |
| 44 | 170 | 70 | 74 | 76 | 76 | 88 |
| 45 | 165 | 69 | 70 | 71 | 71 | 79 |
| 46 | 140 | 66 | 66 | 65 | 68 | 69 |
| 47 | 130 | 63 | 62 | 62 | 65 | 64 |
| 48 | 120 | 51 | 53 | 53 | 54 | 56 |
| 49 | 110 | 40 | 41 | 40 | 42 | 41 |
| 50 | 100 | 14 | 16 | 16 | 14 | 19 |
| 51 | 90 | 9 | 12 | 12 | 11 | 13 |
Table 4: Calculated unscaled frequencies of Bis (thiourea) Cadmium Bromide(BTCB) computed at HF, DFT (B3LYP& B3PW91), CAM - B3LYP & LSDA with3-21G (d, p) basis sets.
| Energy levels | Energy in eV |
|---|---|
| H+8 | 6.903 |
| H+7 | 6.684 |
| H+6 | 6.575 |
| H+5 | 6.258 |
| H+4 | 6.147 |
| H+3 | 6.027 |
| H+2 | 5.971 |
| H+1 | 5.838 |
| H | 5.751 |
| L | 1.734 |
| L-1 | 1.583 |
| L-2 | 0.858 |
| L-3 | 0.446 |
| L-4 | 0.036 |
| L-5 | 0.121 |
| L-6 | 0.420 |
| L-7 | 0.931 |
| L-8 | 1.033 |
| L-9 | 1.414 |
| L-10 | 1.449 |
Table 5: Frontier molecular orbitals with energy levels of Bis (thiourea) Cadmium Bromide (BTCB).
Amino group vibrations: The molecule is populated with couple of thiourea which contains bi NH2 groups. Generally, the amino group is a dominated ligand and make the impression strong in the vibrational pattern. As there are eight N-H bonds, eight vibrational bands of stretching modes are possible. Normally, the primary amines are recognized by strong absorption peaks in the regions of 3450- 3100 cm−1 and 3100-3300 cm−1 due to the asymmetric and symmetric N-H stretching respectively [13,14]. Also the NH2 + asymmetric and symmetric deformation wave numbers are expected to fall in the regions 1660-1610 cm−1 and 1550-1485 cm−1 respectively [15,16]. Also the observed N-H stretching frequencies are found at 3350, 3345, 3320, 3300, 3295, 3290, 3250 and 3245 cm−1. Out of these, the first three bands are assigned to asymmetric vibrations and the rest five bands are assigned to symmetric vibrations. Apart from these assignments, the last two vibrational bands are found to be moved down from the expected region. Due to the dominating character of the NH group, all the stretching vibrations should be observed within the expected region. But this is not so in this case. This is mainly due to the presence of sulphur with chain. The presence of N-H in plane bending vibrations (scissoring) are usually observed in the region 1610-1630 cm−1, rocking vibrations are assigned in the range 1100-1200 cm−1 and the out of plane bending (wagging and twisting) vibrations are normally identified under the region 1150-900 cm-1 [17-19]. In the present compound, the in-plane deformation vibrations are observed at 1610, 1600, 1590, 1565, 1540, 1490, 1470, 1450 and 1420 cm−1. The first two bands are moved up to the higher region and it is cleared that these vibrations are favoured and not affected by the sulphur. The out of plane bending vibrations are observed at 990, 860, 720, 690, 650, 600, 560 and 510 cm−1. Normally, whenever the metal atom coupled with the organic molecule, those normal vibrational modes of the same are suppressed much. The entire out of plane bending vibrations are found out of the expected region. This observation obviously shows that the N-H out of plane vibrations are hindering by metal complex vibrations. The entire out of plane vibrational modes are affected by other substitutions in the chain and this is observed from N-H vibrations.
C-N vibrations: The C-N stretching frequency is rather a tricky assignment since there exists problem in cascading of these frequencies with other vibrations [20]. According to the previous work [21], the C-N stretching vibrations were found in the region 1386-1266 cm−1 for aromatic amines. In this present work, the C-N Stretching vibrations are observed at 1380, 1310, 1260 and 1190 cm−1which is making disagreement with the literature [22] due to the loading of sulphur and metal atoms with the molecule. The C-NH2 in-plane and out-ofplane bending vibrations are appeared at 390 and 380 cm−1 and 170 and 165 cm−1 respectively. These two vibrations are affected much by other vibrations which make disagreement with literature values [23,24]. From these vibrations, it is observed that, the entire vibrations are altered with respect to the metal vibrations, even though the atom is bonded coordinate covalently. So this view ensures that, the metal is strongly bonded with thiourea and the crystal property of the thiourea is enriched by the addition of metal.
C-S vibrations: The rope connection of thio-cyanate complex is linked through the nitrogen atom or the sulphur atom. This bonding can be easily identified intensively by C-S stretching vibration which appeared in the region 730-690 cm−1 [25-27]. In this present case, the C-S stretching vibrations are identified at 1240 cm−1 (medium intensity) and 1210 cm−1 (weak intensity) in IR spectrum. The observed bands are in agreement with the expected range and literature [28]. Usually, the C-S in-plane bending vibrations are observed in the region of 440- 410 cm−1 [28]. In this metal organic compound, the in-plane bending vibrations are found at 360 and 310 cm−1 and the out-of-plane bending vibrations are found at 140 and 130 cm−1. These vibrational bands are pulled down to the lower region in the expected range and are due to the Vander Waals coupling of Cd ion.
Cd-Br and Cd-S vibrations: The BTCB molecule is a metalorganic crystal compound which comprises Cd metal ion linked with bromine atoms by forming coordinate covalent bond. Normally, in cadmium metal complex, the Cd-Br stretching is very important and is usually observed in the region 315-120 cm−1 [29]. In this compound, the coordinate covalent bond stretching vibrations are identified at 500 and 480 cm−1. The Cd-Br in-plane bending vibrational peaks are appeared at 290 and 270 cm−1 and out-of-plane bending vibrational peaks are appeared at 120 and 110 cm−1. It is concluded that, the Cd- Br vibrations are elevated to higher region. This observation clearly shows that the metal-organic inter nuclear distances are made up of coordinate covalent bond and are weak. Usually, these vibrations will not be affected in order to emphasize its uniqueness character.
In BTCB molecule, the organic compound bisthiourea is directly connected through sulphur atom with metal bromide by forming coordinate covalent bond as S-Cd-S. Due to the large force constants and strong covalent bonds, generally, the Cd-S vibrations are pushed to the higher region by organic vibrations. The Cd-S bond is made up of coordinate covalent bond which is a very a weak bond and its vibrations fallback to the Cd-Br vibrations. In this present case, the Cd-S stretching vibrations are observed at 420 and 400 cm−1. The corresponding in-plane bending vibrations are found at 220 and 200 cm−1 and out-of-plane bending vibrations are observed at 100 and 90 cm−1. The entire vibrations of Cd-S are observed in the lower region of the IR spectrum. This observation shows the weak attraction of the bond between the metal and organic compound. Although the bond observed in the present molecule is weak, its chemical properties are good and the present molecule possesses piezoelectric effect.
Frontier molecular analysis
The probable transitions in electronic-vibrational energy levels of frontier molecular orbitals are used to identify the electro-optical properties of the organic compounds. The overlapping of molecular orbitals in bonding and antibonding is used in making the stabilization of orbital [30]. In molecular interaction, there are two important orbitals that involved in interacting with each other. They are HOMO and LUMO. HOMO is the highest energy occupied molecular orbital that represents the ability to donate an electron. LUMO is the lowest energy unoccupied molecular orbital that represents the ability to accept an electron. These orbitals are also called the frontier orbitals. The interaction between them is much stable and is called filled empty interaction. During the interaction, the electron density is generally occupied in the region between two nuclei. The energy of in-phase interaction is greater than the out-of-phase interaction and forms bonding and antibonding molecular orbital.
The 3D view of frontier orbitals in gas is shown in Figure 5. In the figure, the HOMO is mainly localized over the cadmium, Br, N atoms and C-S group in which the two sigma bond interactions are observed over the C-S of thiourea and one delta bond interaction found over cadmium bromide. The N and Br atoms of the molecule are connected by S orbital lobes. However, LUMO is characterized by a charge distribution that connects the cadmium-bromide atoms and C-S bonds in which there are two sigma and one delta bond interactions are identified. From this observation, it is inferred that, the in-phase and out-of-phase interactions are present in HOMO and LUMO respectively. The HOMO→LUMO transition implies that an electron density is transferred between cadmium bromide and thiourea separately. Thus, the obtained transitions in the electron clouds of thiourea and metal complex ensure the occurrence of incorporation of physical and chemical properties. The kubo gap energy of the present material is 4.02 eV, which shows moderate electrical activity and effective optical activity.

Figure 5: Frontier molecular orbital lobe formation.
NMR analysis
Usually, the NMR signals of the compounds explain the chemical environment of the carbons. In this case, the carbons are situated in different environment and proportionately, the chemical properties are obtained alternatively. The chemical shifts of the compound are reported in ppm relative to TMS for 1H and 13C NMR spectra and are presented in Table 6.
| According to TMS B3LYP/ 6-311+G (2d, p) (ppm) |
||||
|---|---|---|---|---|
| Atom position | Gas | DMSO | Chloroform | CCl4 |
| S1 | 187.89 | 256.32 | 240.74 | 221.88 |
| S2 | 185.23 | 250.47 | 235.07 | 217.02 |
| C3 | 166.03 | 175.80 | 173.48 | 170.91 |
| C4 | 168.92 | 177.51 | 175.36 | 173.06 |
| N5 | 94.17 | 93.89 | 94.49 | 94.59 |
| N8 | 84.07 | 89.92 | 88.52 | 86.90 |
| N11 | 85.31 | 91.17 | 89.70 | 88.08 |
| N14 | 92.34 | 93.10 | 93.39 | 93.21 |
| H6 | 1.86 | 2.68 | 2.47 | 2.25 |
| H7 | 2.32 | 2.48 | 2.43 | 2.39 |
| H9 | 1.40 | 2.40 | 2.14 | 1.86 |
| H10 | 1.60 | 2.21 | 2.06 | 1.89 |
| H12 | 1.34 | 2.40 | 2.11 | 1.81 |
| H13 | 1.68 | 2.25 | 2.12 | 1.97 |
| H15 | 1.86 | 2.67 | 2.45 | 2.23 |
| H16 | 2.15 | 2.47 | 2.41 | 2.33 |
| Cd17 | 3759.86 | 3807.27 | 3788.83 | 3776 |
| Br18 | 2845.51 | 2885.67 | 2878.86 | 2870.03 |
| Br19 | 2840.04 | 2945.81 | 2927.03 | 2903.36 |
Table 6: Calculated 1H and 13C NMR chemical shifts of Bis (thiourea) Cadmium Bromide (BTCB).
In the present compound, the metallo-organic compound has been taken for the study in which the molecule contains two carbons along with two amine groups. The 13C NMR chemical shift of such two carbons is greater than 100 ppm, as in the expected regions.
In this case, the chemical shift of C3 is 166.03 ppm and that of C4 is 168.92 ppm. Since both the carbons C3 and C4 have similar groups, the chemical shift is same for both. Due to the migration of double bond from C-S to C-N, the chemical shift of both carbons is very high. The chemical shift of Br (2845.51) is finite and apparently high due to the random breaking of proton shield by the fusing of coordinate covalent bond with organic molecules. The chemical shift of H6, H7, H9, H10, H12, H13, H15 and H16 are calculated as 1.86, 2.32, 1.40, 1.60, 1.34, 1.68, 1.86 and 2.15 ppm respectively. From these result, it is observed that, the chemical shift of H7 and H16 are higher than the rest of other hydrogen atoms in the chain. This is purely due to the extended influence on hydrogen atom by nearby bromine atoms. Hence it is concluded that the chemical property of the metal is directly coupled with organic molecules and this shows that the present metal complex molecule have an additional chemical property.
Optical property analysis
The UV and visible spectrum of the compound in gas and different solvents (DMSO, chloroform and CCl4) are calculated at B3LYP/3-21 G (d, p) level using the TD-DFT approach. The calculated excitation energies, oscillator strength (f), wavelength (λ) and spectral assignments are given in Table 7. The major contributions of the transitions are assigned according to the result of SWizard program [31].
| λ (nm) | E (eV) | ( f ) | Major contribution | Assignment | Region | Bands |
|---|---|---|---|---|---|---|
| Gas | ||||||
| 654.03 | 1.8957 | 0.0061 | H→L | n→σ* | Visible | R-band (German, radikalartig) |
| 562.82 | 2.2029 | 0.0037 | H→L | n→π* | Visible | |
| 557.12 | 2.2254 | 0.0046 | H→L | n→π* | Visible | |
| DMSO | ||||||
| 426.60 | 2.9064 | 0.0084 | H→L | n→π* | Visible | R-band (German, radikalartig) |
| 393.90 | 3.1476 | 0.0045 | H→L | n→π* | UV | |
| 380.92 | 3.2549 | 0.0055 | H→L | n→π* | UV | |
| Chloroform | ||||||
| 472.61 | 2.6234 | 0.0082 | H→L | n→π* | Visible | R-band (German, radikalartig) |
| 439.56 | 2.8206 | 0.0032 | H→L | n→π* | Visible | |
| 401.88 | 3.0851 | 0.0044 | H→L | n→π* | Visible | |
| CCl4 | ||||||
| 528.11 | 2.3477 | 0.0082 | H→L | n→π* | Visible | R-band (German, radikalartig) |
| 493.37 | 2.5130 | 0.0033 | H→L | n→π* | Visible | |
| 445.78 | 2.7813 | 0.0053 | H→L | n→π* | Visible | |
Table 7: Theoretical electronic absorption spectra of Bis (thiourea) Cadmium Bromide (BTCB) (absorption wavelength γ (nm), excitation energies E (eV) and oscillator strengths (f)) using TD-DFT/B3LYP/3-21G (d, p) method.
According to TD-DFT calculations, the overall transitions belong to quartz UV region. In gas phase, the strong transition is observed at 654 nm and its oscillator strength is f=0.0061 with energy gap of 1.89 eV. Also in this phase, two more strong transitions are observed at 562 and 557 nm and their corresponding oscillator strengths are f= 0.0037 and 0.0046 with energy gap of 2.20 and 2.22 eV. These transitions are indicated as n → σ* which belongs to the visible region. In this, the band is designated as R-band (German, Radikalartig) which is attributed to the above said transition of chain of chromophoric groups, such as Cadmium bromide group.
From TD-DFT calculations, the following features are inferred. Their molar absorptivities are low. They transferred from hypsochromic to bathochromic shift. In this case, the solvent effect is inactive. The simulated UV-Visible spectra in gas and solvent phase are shown in Figure 6.

Figure 6: Simulated UV-Visible spectra of BTCB.
In the case of DMSO solvent, the calculated absorption spectrum indicates that, the maximum absorption wavelength corresponds to the electronic transition and it undergoes from the HOMO+1 to LUMO-1 with maximum contribution. The Frontier molecular orbital diagram is presented in the Figure 7. Here, the chromophores are cadmium bromide group, the crystal properties are increased in the present compound. In the case of DMSO solvent, strong transitions are observed at 426, 393 and 380 nm and their corresponding oscillator strengths are f=0.0084, 0.0045 and 0.0055 with maximum energy gap 3.25 eV. They are denoted as n → π* transition and belongs to the visible region. One of the electronic transitions is observed at IR region. Hence from gas to solvent, the electronic transitions retained at the visible region. This observation clearly indicates that, the present molecule has visible active and it has very high optical properties. Moreover, the optical band gap is calculated as 3.25 eV and this view clearly ensure that the present compound BTCB possess LO as well as NLO properties.

Figure 7: Successive Homo-Lumo of BTCB.
The chemical hardness and potential, electro negativity and Electrophilicity index are calculated and their values are shown in Table 8. The chemical hardness is a property which has good chemical stability. For the present compound BTCB, the chemical hardness is calculated as 2.00 and so it has high chemical stability. The chemical stability and metal character of the compound is enhanced by substituting the Cd-Br group.
| Parameters | TD-DFT/B3LYP/3-21G |
|---|---|
| Etotal (Hartree) | -11643.95 |
| EHOMO (eV) | 5.7513 |
| ELUMO (eV) | 1.7341 |
| ΔEHOMO-LUMO gap (eV) | 4.0172 |
| EHOMO-1 (eV) | 0.0874 |
| ELUMO+1 (eV) | 0.1510 |
| ΔEHOMO-1-LUMO+1 gap (eV) | 0.0636 |
| Chemical hardness (h) | 2.0086 |
| Electronegativity (χ) | 3.7427 |
| Chemical potential (μ) | 2.0086 |
| Chemical softness(S) | 0.2489 |
| Electrophilicity index (ω) | 3.4869 |
| Dipole moment | 1.9775 |
Table 8: Calculated energies values, chemical hardness, electro negativity, Chemical potential, Electrophilicity index of Bisthiourea Cadmium Bromide (BTCB).
Also, the electro negativity of the compound is calculated as 3.74 and this property shows that the chemical bonds in the present molecule will be changed from covalent to ionic. The bonding nature of the present compound is rehabilitated to rich ionic property and this is due to the addition of Cd atom.
Electrophilicity index is a factor which is used to measure the energy lowering due to maximal electron flow between donor [HOMO] and acceptor [LUMO]. From the Table 8, it is found that the Electrophilicity index is 3.48 which is high and this value ensures that the strong energy transformation is taking place between HOMO+1 and LUMO-1. One more important electronic property of the molecule is its dipole moment. Whenever the molecules possess large dipole moment, the intermolecular interactions are very strong. The dipole moment value for the present compound BTCB is calculated as 1.97 Debye. This value is temperate due to the presence of coordinate covalent bond. Hence, the present molecule possesses strong intermolecular interactions.
Molecular Electrostatic Potential (MEP) analysis
The molecular electrostatic potentials have been used for interpreting and predicting the reactive behaviour of a wide variety of chemical systems in both electrophilic and nucleophilic reactions, the study of biological recognition processes and hydrogen bonding interactions [32,33]. Molecular electrostatic potential (MEP) at a point around a molecule gives an indication of the net electrostatic effect produced at that point by the total charge distribution (electron+nuclei) of the molecule and correlates with dipole moments, electro negatively, partial charges and chemical activity of the molecules. It provides a visual method to understand the relative polarity of the molecule. An electron density iso-surface mapped with electrostatic potential surface depicts the size, shape, charge density and site of chemical reactivity of the molecules. The different values of the electrostatic potential at the surface are represented by different colours; red represents regions of most negative electrostatic potential, blue represents regions of most positive electrostatic potential and green represents regions of zero potential. Potential increases in the order of red

Figure 8: MEP map Diagram with isosurface of BTCB.
From the MEP map of the present compound, the negative regions are mainly localized on cadmium bromide and sulphur atoms. Due to the presence of maximum positive region on the H of NH2 groups, there exists the possibility of nucleophilic attack in the region. The above calculated results inferred that the metal atoms coupled strongly in the position of organic lattice region.
Polarizability and first order hyperpolarizability calculations
Using DFT-B3LYP method and 3-21 G (d, p) basis set, based on the finite-field approach, NLO properties, binding properties, the polarizabilities and first order hyperpolarizabilities of the title molecule are calculated. These parameters are used to find the relationships between the structures of the molecule.
The mean polarizability (α), anisotropy of polarizability (Δα) and the average value of the first hyperpolarizability 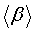
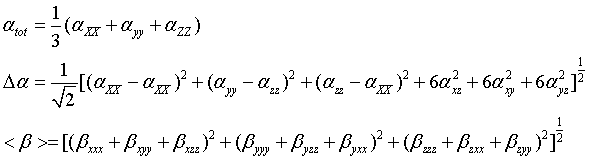
where αxx, αxy, αyy, αxz, αyz, αzz and βxxx, βxxy, βxyy, βyyy, βxxz, βxyz, βyyz, βxzz, βyzz, βzzz are the Polarizability and hyperpolarizability tensors. These are obtained from the output file of Polarizability and hyperpolarizability calculations. The values α and β of Gaussian output are in atomic units. They have been converted into electronic units.
For ‘α’, 1 a.u.=0.1482 × 10−24 esu,
For ‘β’, 1 a.u.=8.6393 × 10−33 esu
A molecule whose dipole moment, molecular polarizability and first hyperpolarizability values are high will have high active NLO properties.
The first hyperpolarizability and their components βx, βy and βz of the present molecule along with related properties such as dipole moment, average polarizability, anisotropy of polarizability are given in Table 9. In this, the value of dipole moment can be calculated as 4.33 Debye. The dipole moment in the component of μz is observed as 4.33 D and this value is high. The lowest value of the dipole moment of the molecule compound is μy component and it is calculated as-0.0089 D. The polarization in different coordinate in the material tuned the optical energy that enters. The calculated value of α=153.47 × 10-24 esu and Δα=115.41 × 10−24 esu. In the present molecule, the Polarizability is found to be large in amount. This high value of Polarizability shows that the present molecule is rich in NLO property. So the present metallo-organic compound is clearly optically active. The magnitude of the molecular hyperpolarizability β, is one of the important key factors in a NLO system. Because, hyperpolarizability of a system induces optical modulation inside the material and it also stimulating second order harmonic generation in the lattice site. Using (B3LYP/3-21G(d, p)) method, first hyperpolarizability value is calculated as β=-138.83 × 10−30 esu. The high value of hyperpolarizability of the title compound emphasize the generation of the second order harmonic generation with more amplitude. So, the present compound is able to prepare the NLO crystals for enriched electronic applications.
| Parameter | LSDA 3-21G (d, p)(a.u.) | HF 3-21G (d, p)(a.u.) | Parameter | LSDA 3-21G (d, p)(a.u.) | HF 3-21G (d, p)(a.u.) |
|---|---|---|---|---|---|
| αxx | -42.9340 | -25.6242 | βxxx | 0.0349 | 0.1100 |
| αxy | 14.7379 | 22.9900 | βxxy | -0.0424 | -0.1562 |
| αyy | -136.9602 | -139.8447 | βxyy | 0.0022 | -0.0581 |
| αxz | -0.0057 | 0.0109 | βyyy | 0.0047 | -0.0409 |
| αyz | 0.0025 | -0.0081 | βxxz | -55.9116 | 59.7263 |
| αzz | -128.8208 | -138.6108 | βxyz | 24.9887 | 27.4902 |
| αtot | 183.312 | 153.476 | βyyz | -2.6321 | 10.8741 |
| Δα | 283.314 | 115.416 | βxzz | -0.0132 | 0.0178 |
| μx | 0.0004 | 0.0021 | βyzz | -0.0041 | 0.0413 |
| μy | -0.0029 | -0.0089 | βzzz | -67.2794 | -76.2617 |
| μz | -1.9775 | 4.3309 | βtot | -2874.4497 | -138.8311 |
| μ | 1.9775 | 4.3309 |
Table 9: The dipole moments μ (D), the polarizability α (a.u.), the averagepolarizability αo (esu), the anisotropy of the polarizability Δα (esu), and the firsthyperpolarizability β (esu) of Bis (thiourea) Cadmium Bromide (BTCB) computedat HF & LSDA with 3-21G (d, p) basis sets.
| T(K) | 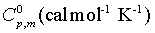 |
 |
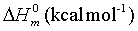 |
Observed Peak Endothermic signal |
|---|---|---|---|---|
| 100.00 | 404.94 | 133.84 | 9.12 | - |
| 200.00 | 515.37 | 189.18 | 25.37 | 199.08 |
| 298.15 | 599.21 | 231.74 | 46.12 | 247.66 |
| 300.00 | 600.64 | 232.44 | 46.55 | - |
| 400.00 | 672.28 | 265.82 | 71.53 | - |
| 500.00 | 734.53 | 292.01 | 99.47 | - |
| 600.00 | 789.68 | 312.88 | 129.76 | - |
| 700.00 | 839.23 | 329.92 | 161.92 | - |
| 800.00 | 884.25 | 344.20 | 195.65 | - |
| 900.00 | 925.51 | 356.40 | 230.70 | - |
| 1000.00 | 963.62 | 366.95 | 266.87 | - |
Table 10: Thermodynamic parameters at different temperatures at the B3LYP/3-21G (d, p) level for Bis (thiourea) Cadmium Bromide (BTCB).
Thermodynamic properties analysis
Thermo dynamic properties provide the necessary information regarding the chemical reactivity. Moreover it is used to discuss the existence and alternation of thermodynamic parameters of the present compound since the molecule is a metal-organic substance. The values of some thermodynamic parameters such as standard heat capacities (C0 p,m), standard entropies (S0 m) and standard enthalpy changes (ΔH0m) of title molecule by B3LYP/3-21 G (d, p) method are listed in Table 10. On the basis of vibrational analysis, these values were obtained from the theoretical harmonic frequencies. From Table 10, it can be observed that these thermodynamic functions are increased with temperature ranging from 100 to 1000 K due to the fact that the molecular vibrational intensities increase with temperature. The correlation graph between heat capacities, entropies, enthalpy changes and temperatures were shown in Figure 9.

Figure 9: Thermodynamical parameters variation diagram.
From this observation, it is clear that, the dissociation of atoms related to the temperature is increased up to 1000 K and the molecule has positive entropy-coefficient. In the case of thermodynamical analysis of the molecule, the enthalpy of a system due to the production of metal ion and organic interactions is found to be increased with consecutive saturation between the successive temperatures (e.g., 300 K-400 K). At low temperature, it is found that, the specific heat capacity of the present compound falls down rapidly and obeys the Debye T3 law.
TGA/DTA analysis
The Thermo gravimetric analysis and differential thermal analysis for BTCB have been performed at a heating rate of 10°C/ min in nitrogen and are reported in Figure 10. In the analytical graph, there are two endothermic peaks which are observed at 199.08°C and 247.66°C. The peaks are observed due to the liberation of bromide and cadmium atoms from the crystal which are due to the weak coordination covalent interaction with thiourea. The formation of the metal complex with thiourea in the inner coordination sphere indicates greater thermal stability of the crystal [34]. The TGA analysis curves show that the occurring of weight loss which is about 81.01% in the temperature range 178-602°C and this is due to the detachment of metal bromide.

Figure 10: TGA/DTA curve for BTCB.
Natural Bond Orbital (NBO) analysis
Using DFT/B3LYP level, the Natural Bond Orbital (NBO) calculations are performed in order to understand various second order interactions between the filled orbital of base system and vacant orbital of ligand system and vice versa, which is a measure of the intermolecular delocalization or hyper-conjugation. The NBO investigation facilitates the most accurate possible natural Lewis structure with the orientation of electron density. The useful aspect of the NBO method is that it gives information about interactions of both filled and virtual orbital interactions. In this case, the second-order Fock-matrix is carried out to evaluate the donor-acceptor interactions in the NBO basis. For each donor (i) and acceptor (j), the stabilization energy [E(2)] associated with the delocalization i → j is determined as
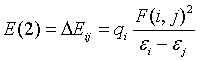
where qi is the donor orbital occupancy; εi, and εj are the diagonal elements (orbital energies) and F (i, j) is the off-diagonal NBO Fock-matrix clement. In NBO analysis, large value of E(2) shows the intensive interaction between electron-donors and electronacceptors and greater the extent of conjugation of the whole system, the possible intensive interactions are given in Table 11. The secondorder perturbation theory analysis of Fock-matrix in NBO basis shows strong intra-molecular hyper-conjugative interactions of σ and π electrons. The intra molecular hyper-conjugative interactions are formed by the orbital overlap between the thiourea segments and metal-bromide. Though the metal bromide is connected symmetrically with thiourea by coordinate covalent bond, the strong intra-molecular hyper-conjugative interaction is taking place between the lone pair of S1 and σ of Cd17-Br18 that weakens the respective bonds leading to the stabilization of 6.07 kJ mol-1. The another intra molecular hyperconjugative interaction is formed by the orbitals of S2-C4 and Cd17- Br (18 and 19) overlap between thiourea and cadmium atom and bond orbital by spending 6.05 kJ mol-1 which results in ICT causing stabilization of metal organic system. Simultaneously, the interaction is taking place between S2-C4 and N11-H (12 and 13) by spending 3.30 kJ mol-1. This view shows the mutual coupling of thiourea and Cd in left moiety. The same amount of energy has been spent to form the interaction between C-S and Cd in right moiety of the total system. Thus, a strong intra molecular hyper-conjugative interaction has occurred between metal atom and thiourea. From these interactions, it is notable that, in the case of Cd-Br, the electron density move towards Br from Cd by making it positive. In the case of C-S and N-H, the electron clouds are pulled by C and N by leaving S neutral. The increased electron density at the Br and N atoms leads to the elongation of respective bond length and a lowering of the corresponding stretching modes. The electron density (ED) is transferred from the n(Br) to the anti-bonding of σ* and π* orbital of the BTCB explaining both the elongation and the red shift. This view is strongly validated by Mulliken charge analysis.
| Donor (i) | Type of bond | Occupancy | Acceptor (j) | Type of bond | E(2) kcal/mol | E(j) - E(i) a.u. | F(i, j) a.u. |
|---|---|---|---|---|---|---|---|
| S1 | σ | 1.999 | Cd17 | σ* | 20.58 | 88.22 | 0.159 |
| S1 | σ | 1.999 | Cd17-Br18 | σ* | 6.07 | 7.70 | 0.04 |
| S1 | σ | 1.999 | Cd17-Br19 | σ* | 5.44 | 5.88 | 0.020 |
| S1 | σ | 1.999 | C3-N14 | σ* | 23.63 | 0.21 | 0.067 |
| S1-C3 | σ | 1.977 | N8 | σ* | 1.15 | 2.14 | 0.045 |
| S1-C3 | σ | 1.977 | N14 | σ* | 1.16 | 2.16 | 0.045 |
| S1-C3 | σ | 1.977 | N8-H9 | σ* | 3.29 | 1.01 | 0.052 |
| S1-C3 | σ | 1.977 | N14-H15 | σ* | 3.24 | 1.02 | 0.051 |
| S1-C3 | σ | 1.977 | Cd17-Br18 | σ* | 0.09 | 0.73 | 0.007 |
| S1-C3 | σ | 1.977 | Cd17 | σ* | 1.84 | 0.71 | 0.034 |
| S2 | σ | 1.999 | C4-N5 | σ* | 23.63 | 0.21 | 0.066 |
| S2 | σ | 1.999 | Cd17 | σ* | 20.57 | 0.32 | 0.073 |
| S2 | σ | 1.999 | Cd17-Br18 | σ* | 5.46 | 0.34 | 0.040 |
| S2 | σ | 1.999 | Cd17-Br19 | σ* | 6.05 | 0.34 | 0.042 |
| S2-C4 | σ | 1.977 | Cd17 | σ* | 0.73 | 0.71 | 0.022 |
| S2-C4 | σ | 1.977 | N5-H6 | σ* | 3.24 | 1.02 | 0.051 |
| S2-C4 | σ | 1.977 | N11-H12 | σ* | 3.30 | 1.01 | 0.052 |
| C3-N14 | π | 0.498 | C3 | π* | 0.55 | 1.06 | 0.043 |
| C3-N8 | σ | 1.994 | N8-H9 | σ* | 0.60 | 1.26 | 0.025 |
| C3-N14 | π | 1.996 | C3-N14 | π* | 4.79 | 0.34 | 0.041 |
| C4 | σ | 1.999 | Cd17 | σ* | 0.05 | 10.20 | 0.022 |
| C4-N5 | π | 1.996 | Cd17 | π* | 0.37 | 0.44 | 0.012 |
| C4-N5 | π | 1.996 | C4-N5 | π* | 7.76 | 0.48 | 0.103 |
| C4-N11 | σ | 1.994 | Cd17 | σ* | 0.15 | 0.96 | 0.011 |
| N5-H6 | σ | 1.985 | S2-C4 | σ* | 4.45 | 0.86 | 0.055 |
| N5-H7 | σ | 1.986 | C4-N11 | σ* | 4.11 | 1.09 | 0.060 |
| N5-H7 | σ | 1.986 | Cd17 | σ* | 0.54 | 0.73 | 0.019 |
| N5-H7 | σ | 1.986 | Cd17-Br18 | σ* | 0.22 | 0.75 | 0.012 |
| N5-H7 | σ | 1.986 | Cd17-Br19 | σ* | 0.07 | 0.75 | 0.007 |
| N8 | σ | 1.998 | C3-N14 | σ* | 75.42 | 0.23 | 0.123 |
| N8-H9 | σ | 1.988 | C3 | σ* | 2.89 | 1.41 | 0.057 |
| N8-H10 | σ | 1.990 | C3 | σ* | 2.60 | 1.58 | 0.057 |
| N11 | σ | 1.998 | C4-N5 | σ* | 75.89 | 0.23 | 0.123 |
| N11-H 13 | σ | 1.990 | C 4 - N 5 | σ* | 3.72 | 1.12 | 0.058 |
| N11-H13 | σ | 1.990 | Cd17 | σ* | 0.05 | 0.75 | 0.006 |
| N14 | σ | 1.998 | C3 | σ* | 5.87 | 14.81 | 0.264 |
| N14-H15 | σ | 1.985 | C3 | σ* | 2.49 | 1.40 | 0.053 |
| N14-H16 | σ | 1.986 | C3 | σ* | 1.48 | 1.38 | 0.040 |
| N14-H16 | σ | 1.986 | Cd17-Br18 | σ* | 0.07 | 0.75 | 0.007 |
| N14-H16 | σ | 1.986 | Cd17-Br19 | σ* | 0.22 | 0.75 | 0.012 |
| Cd17-Br18 | σ | 1.970 | H16 | σ* | 0.06 | 1.34 | 0.008 |
| Cd17-Br18 | σ | 1.970 | S1-C3 | σ* | 0.05 | 0.60 | 0.005 |
| Cd17-Br19 | σ | 1.970 | N14 | σ* | 0.05 | 1.93 | 0.009 |
| Cd17-Br19 | σ | 1.970 | H16 | σ* | 0.34 | 1.34 | 0.019 |
| Cd17-Br19 | σ | 1.970 | N14-H16 | σ* | 4.39 | 0.79 | 0.053 |
| Br18 | σ | 2.000 | N5-H7 | σ* | 20.03 | 0.58 | 0.097 |
| Br19 | σ | 2.000 | N14-H16 | σ* | 20.00 | 0.58 | 0.097 |
| Cd17 | σ | 1.999 | Cd17-Br18 | σ* | 19.82 | 0.02 | 0.042 |
| Cd17 | σ | 1.999 | Cd17-Br19 | σ* | 19.93 | 0.02 | 0.042 |
Table 11: Second order perturbation theory analysis of Fock matrix in NBO basis corresponding to the intra-molecular bonds of the Bis (thiourea) Cadmium Bromide(BTCB).
FT-IR, FT-Raman, UV, NMR and quantum chemical calculation studies have been performed on Bis Thiourea Cadmium Bromide (BTCB) to identify its structural and spectroscopic properties. A complete vibrational analysis of BTCB was performed with HF and DFT method using 3-21G (d, p) basis set. From the UVVisible spectra, it is monitored that, the entire electronic transitions shifted bathochromically due to the substitutional effect. From the Mulliken charge analysis, the entire charge levels of the molecule are altered on par with due to the substitution. The electrical, optical and bio-molecular properties are profoundly investigated using frontier molecular orbital. From NMR analysis, it is concluded that the chemical property of the metal is directly coupled with organic molecules and this shows that the present metal complex molecule have an additional chemical property. The MEP map is performed and from this, the change of the chemical properties of the compound is also discussed. The TGA/DTA curves recorded for the crystal confirmed its thermal stability. The correlations between the statistical thermodynamics and temperature are also obtained. It is seen that the heat capacities, entropies and enthalpies increase with the increasing temperature owing to the intensities of the molecular vibrations increase with increasing temperature. Furthermore, the average Polarizability, the first order hyperpolarizability and total dipole moment of title molecule have been calculated and the results are discussed. These results indicate that the title compound is a good candidate of nonlinear optical materials. In NBO analysis, both filled and virtual orbital interactions are discussed.