Journal of Antivirals & Antiretrovirals
Open Access
ISSN: 1948-5964
ISSN: 1948-5964
Research Article - (2016) Volume 8, Issue 2
Oseltamivir and zanamivir are the commercially successful neuraminidase (NA) inhibitors approved for treatment of influenza, caused by highly pathogenic H1N1 influenza virus. Being transition state analogs of sialic acid, an endogenous ligand of NA, many scaffolds were developed based on similar structural features. But high rate of mutation in H1N1 necessitates extra search of a novel drug which can withstand drug resistance. Considering this we have developed a known naturally occurring scaffold chalcone, which is not a transition state analog, by incorporating various substituents based on their electronic and steric properties. With the help of computational drug design few promising molecules are screened and synthesized further. All the compounds under study showed different mode of binding in the active cavity. The inhibition of cytopathic effect of H1N1 by synthesized derivatives has been evaluated by in vitro cell based technique using oseltamivir as a standard. To gain insight into molecular mechanism of interaction of the derivatives with H1N1-NA, we have investigated the effect of these molecules on thermotropic properties and organization of membrane bilayer prepared from lipids using nuclear magnetic resonance (NMR) and differential scanning calorimetric (DSC) techniques. The compounds under study point towards stabilization of the membrane bilayer as expected for antiviral activity. Almost all derivatives showed activity parallel to the standard. Among all derivative ChmN showed highest activity at 0.67 nM whereas lowest activity was displayed by derivative ChmM at 10.35 nM. Derivative ChpH showed activity almost equivalent to oseltamivir.
<Keywords: Neuraminidase inhibitors; H1N1; Chalcone; Molecular docking; NMR; DSC; Antiviral activity
The highly pathogenic H1N1 influenza virus remains a constant threat due to its high rate of mutability. The currently available antiviral therapy such as M2 inhibitors and Neuraminidase (NA) inhibitors may show resistance if exposed for prolong period of time. Oseltamivir and zanamivir are the commercially successful NA inhibitors approved for treatment of influenza A and B infections [1]. It has been reported that influenza virus with NA subtype N1 would be resistant to oseltamivir due to side chains of Glu119 and Asp151 may not have the precise alignment required to bind oseltamivir tightly [2]. Similarly, zanamivir resistance has been associated with mutation of Gln136 to Lys which breaks down the bonding network formed with Asp151 and Arg156 leading to increased mobility of both groups [3]. Earlier studies in the development of NA inhibitors were based on synthesis of transition analogs of sialic acid which is an endogenous ligand [4]. Many scaffolds were developed based on similar structural features [5].
We have focused our study to develop a resistance free antiviral drug using a different approach by selecting a scaffold which will neither have structural similarity to sialic acid nor to the currently popular NA inhibitors. In the search we have selected chalcone, a phytoconstituent derived from roots of Glycyrrhiza, which have been reported to be active against H1N1-NA. However they show lesser NA inhibitory activity as compared to commercial drugs [6]. We have further developed this scaffold based on electronic and steric properties with substituents on benzylidene ring of chalcone moiety (1, 3-diphenyl-prop-3-ene- 1-one) keeping 2’- Hydroxy group at phenyl moiety constant. The substituted analogs are 2/3/4-chloro, 2/3/4 -methoxy, 2/3/4-hydroxy and 2/3/4-nitro chalcones (Figure 1). In addition we have compared the results with quercetin, a naturally occurring non competitive inhibitor of H1N1, and flavone moiety. All these molecules are not transition state analogues.

Figure 1: Molecular structure of chalcone (1, 3-diphenylprop-2-en-1-one) and its derivatives.
We have used molecular docking, a computational tool to investigate the ligand - H1N1 NA interactions of above chalcone derivatives. To compare the effect of these groups on the binding of chalcone derivatives to the H1N1-NA enzyme, the parent compound (zanamivir) and substituted chalcone derivatives were docked into the H1N1-NA active site and analyzed for their binding energies. The activity of these synthesized derivatives was also evaluated in vitro using cell based antiviral studies [7]. Based on the docking results and antiviral activity we have selected three representative analogues 2’- Hydroxy- 4- methoxychalcone (ChpM), 2’- Hydroxy- 4- hydroxychalcone (ChpH), 2’- Hydroxy- 4- nitrochalcone (ChpN) for further studies.
There have been reports that NA inhibitors affect the entry of the virus into the immune cells by interfering with cell-cell fusion [8-12]. This fact of antiviral effect observed with membrane stabilizing effect can be investigated by using model membrane bilayer prepared from a variety of synthetic and natural lipids. Thus in order to gain insight into whether our synthesized derivatives acts like NA inhibitors by interfering with cell-cell fusion, we have investigated the effect of these molecules on thermotropic properties and organization of membrane bilayer prepared from lipids using multinuclear nuclear magnetic resonance (NMR) and differential scanning calorimetric (DSC) techniques. These studies can provide insight into molecular mechanism of interaction of the derivatives with H1N1-NA in order to develop a novel resistance free antiviral drug.
Material
2-Hydroxy acetophenone and various substituted benzaldehydes were purchased from S D fine-chem Ltd., India. L-a-Diapalmitoyl phosphatidyl choline (DPPC) was purchased from Sigma Chemicals Co., U.S.A. All other solvents used for synthesis were of LR grade. Oseltamivir was a gift sample from Cipla Ltd., India. Standard H1N1 virus obtained from National Institute of Virology, Pune, India. Madin- Darby Cannine Kindey (MDCK) cells obtained from Haffkine Institute, Mumbai, India.
Methods
Computational studies: Computational studies were carried out with the modeling package Discovery Studio (DS) 3.1 [13] running on a Red Hat Enterprise Linux WS Workstation. Docking studies were carried out with GOLD v 5.0.1 [14] running on a separate Red Hat Enterprise Linux WS Workstation.
Preparation of enzyme and ligand for docking: The crystal structure of the enzyme H1N1-NA in complex with zanamivir was taken from the protein data bank (PDB ID: 3B7E) [15]. The enzyme is a dimer; however for docking studies only monomeric unit was used. All the water molecules were deleted and hydrogen atoms were added at pH 7. The system was refined using the CHARMm forcefield [16] with the backbone atoms tethered by a force constant of 10 kcal/mol/Å, to a gradient of 0.01 kcal/mol/Å. The zanamivir structure was energy minimized using the “Smart Minimizer” method in Discovery Studio 3.1 that performs steepest descents and conjugate gradients with the CHARMm force field to a gradient of 0.001 kcal/mol/A.
Docking studies: The following parameters in GOLD were permitted to change during the docking runs:(a) the dihedral angles of the inhibitors (b) the inhibitors’ ring geometries (flipping ring corners) (c) the dihedral angles of enzyme OH and NH2 groups and (d) mappings of the H-bonds between the inhibitor and enzyme. At the start of a docking run, all these variables were randomized. Docking was carried out for 20 genetic algorithm (GA) runs, which was found sufficient to reproduce the binding pose of zanamivir in 3B7E. Most of the other GA parameters like population size and the genetic operators were left at their default values. The binding site was defined as a sphere of 10 Å radius around the inhibitor. The docking protocol was validated by the reproduction of the binding pose of zanamivir in 3B7E. This protocol was then used to dock the other synthesized drugs, oseltamivir, and sialic acid to H1N1 to determine their preferred binding orientations. The docked poses were scored using GoldScore. The GOLD fitness function is made up of four components protein-ligand hydrogen bond energy, protein-ligand vander Waals energy (external vdW), ligand internal vdW energy and ligand torsional strain energy. Poses were segregated on the basis of GoldScore.
Synthesis of derivatives of chalcones: We have carried out synthesis of chalcone derivatives based on Claisen-Schmidt condensation reaction [17]. The scheme of overall synthesis of various chalcone derivatives is mentioned in Figure 2. Equimolar quantities of 2-Hydroxy acetophenone and substituted benzaldehyde were dissolved in absolute ethanol. Sodium hydroxide solution (40%) was added to this solution while the temperature was maintained below 10°C throughout. The resulting mixture was stirred at room temperature for 4 h and allowed to stand for 12 h. The precipitate obtained was neutralised with conc. HCl, filtered and recrystallized using alcohol. Completion of reaction was monitored by TLC using n-hexane: ethyl acetate (3:2). The structure and purity of the starting materials and final compounds were confirmed by physical constants, TLC and spectral techniques like IR and NMR spectroscopy (supplementary Table 1).

Figure 2: Synthesis scheme of chalcone and derivatives.
| Amino acid residue | Zanamivir | Oseltamivir | Sialic acid |
|---|---|---|---|
| H1N1-NA | |||
| Arg118 | HO-C=O...HN | NH2..O=C | HO-C=O..HN |
| Glu119 | - | NH..O=C | C-OH..O=C |
| Asp151 | NH2..O=C | - | - |
| Arg152 | - | C=O..H2N | HO-C=O..HN |
| Arg156 | - | - | - |
| Trp178 | NH2..O=C | - | - |
| Ser179 | - | - | - |
| Arg192 | - | - | HO-C=O..HN |
| HO-C=O..HN | |||
| Arg224 | - | - | - |
| Glu227 | NH2..O=C | - | - |
| Tyr247 | - | - | - |
| Glu276 | C-OH..O=C | - | - |
| C-OH..O=C | |||
| Glu277 | - | - | - |
| Arg292 | - | - | - |
| Asn294 | - | - | - |
| Arg371 | O=C-OH..HO | O=C-OH..HN | O=C-OH..HN |
| HO-C=O..HN | |||
| Tyr406 | NH2..O=C | HO-C=O..HN | - |
| HO-C=O..HN | |||
| NH..O=C | |||
| No of H-Bonds | 10 | 5 | 7 |
Atoms involved in hydrogen bonding from ligand (row) to amino acid residue of H1N1-NA (column) are underlined. Last row shows number of hydrogen bond observed in case of each ligands.
Table 1: Results of docking studies showing intermolecular hydrogen bonds of oseltamivir, zanamivir and endogenous ligand sialic acid with H1N1-NA.
Sample preparation for NMR and DSC experiments: Multilamellar vesicles (MLV) were prepared by the standard procedure [18]. Where in the desired quantity of DPPC and chalcone derivatives was dissolved in chloroform. The solvent was then evaporated with a stream of nitrogen so as to deposit a lipid film on the walls of the container. The last traces of the solvent were removed with freeze drying for a period of 1 h. The lipid film was hydrated with the required amount of D2O; this was then incubated in a water bath at 50°C with repeated vortexing. The lipid concentrations were maintained at 100 mM for the NMR and 50 mM for the DSC experiments. Unilamellar vesicles (ULV) were prepared by sonicating the above dispersions with a Branson sonicator (Model 450) at 50% duty cycles till the solution was optically clear.
NMR experiments: NMR experiments were recorded on a BRUKER AVANCE 500 MHz and 700 MHz NMR spectrometer. In 1D proton NMR, 64 scans were recorded. Resonance assignments were carried out using 1D proton NMR and 2D COSY (correlation spectroscopy) spectra which were recorded using standard pulse program at 300 ms mixing time [19-21]. 31P and 13C NMR experiments were carried out with a relaxation delay of 1 s and broadband proton decoupling. The NMR data was processed with Topspin 2.1. Purity of the derivatives was checked from the NMR assignments.
DSC experiments: DSC measurements were carried out on the differential scanning calorimeter VP-DSC (MicroCal, Northampton MA, USA). The samples were degassed under vacuum before being loaded into the reference and sample cells. A scan rate of 10°C/h was employed. Data was analyzed with the software ORIGIN provided by MicroCal. All the experiments were carried out in the temperature range 20°C to 60°C. Repeated scans (n=3) for the same sample were generally superimposable [22].
Determination of MLV-drug (chalcone derivatives) binding: Binding constants were determined by the centrifugation method [23]. Optical density of a 100 µM solution of the drug molecule was measured with the help of UV spectrophotometer (?max 220-400 nm). MLVs were prepared by varying lipid concentration systematically from 0.25 mg/2.0 ml to 2.0 mg/2.0 ml and fixed drug concentration of 100 µM (a drug:lipid ratio 1:2.5 to 1:20). The resulting solutions were incubated for 2 h and subsequently transferred into ultracentrifuge tubes. Separation of liposomes from the aqueous phase was achieved by centrifugation at 15000 rpm for 5 h. The drug concentration in the supernatant was determined by measuring the optical density of the drug molecules and the amount of drug bound to liposomes was determined from the difference. The drug-liposome apparent binding constant (K) was analyzed using the double reciprocal plot. A plot of 1/ (fraction bound) vs. 1/[lipid concentration] yields a straight line with slope 1/K.
AlogP: Hydrophobicity/hydrophilicity of the drug influences the behaviour of a molecule in a living organism, affecting its bioavailability, transport, reactivity, toxicity, metabolic stability and many other properties. We have therefore calculated the AlogP for chalcone derivatives. Calculations were done in DS 3.1 software package using ‘Calculate Molecular Properties’ protocol. AlogP values are as follows ChpM (3.44), ChpH (3.21), ChpN (3.35). AlogP values indicate extent of lipophilicity is in the order ChpM>ChpN>ChpH.
Antiviral evaluation
Cells and virus: Madin-Darby canine kidney (MDCK) cells were grown in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) and 1% Penstrep (Penicillin and Streptomycin). The influenza virus strain A/Pune isolate/2009 (H1N1) was propagated in MDCK cells in the presence of 10 µg/mL trypsin.
Cytotoxicty studies: MDCK cells were grown in 96 well plates for 24 h. The plates were replaced with media containing serially diluted compounds (10 fold dilutions). After 16 h of incubation, the medium was removed and 100 µL MTT (3-(4, 5-dimethylthiozol-2-yl)-3, 5-dipheryl tetrazolium bromide, solution was added to each well and incubated at 37°C for 4 h. After removal of supernatant, 100 µL of Dimethylsulfoxide (DMSO) was added to dissolve formazan crystals. Absorbance was measured at 540 nm in a microplate reader [24]. Cytotoxicity was expressed as the 50% cell-inhibitory concentration (CC50).
Cytopathic effect (CPE) inhibition assay: The virus (100 µl) was inoculated onto near confluent MDCK cell monolayers for 1 h in 24-well plates at 37°C under 5% CO2 atmosphere. The solution was removed and the cells replaced with MEM containing 10 µg/mL trypsin and candidate compounds at different concentration. The cultures were incubated for 3–4 days at 37°C under 5% CO2 atmosphere until the cells in the infected and untreated control well showed complete viral CPE. All compounds were assayed for virus inhibition in duplicate [24]. After 3-4 days, the supernatant from each well was removed and tested for reduction in heamagglutinition (HA) titre of virus as compared to HA titre of virus in untreated control well (virus control). The HA titre was determined by means of HA assay.
Heamagglutination assay: Serial two-fold dilutions of supernatant of infected cells (100 µl) were prepared using PBS in a 96-well plate. A 50 µl of 0.75% guinea pig red blood cells was added to each well. After 30 min incubation at 4°C followed by 30 min incubation at room temperature, heamagglutination (uniform reddish colour across the well) and precipitation of red blood cells (dot in the centre of well) was observed.
Docking
The H1N1-NA active site consists of four well-conserved binding sites with 11 functional residues which participate in the catalytic reaction [25]. These are the positively charged site-1 (Arg118, Arg292 and Arg371), the negatively charged site-2 (Glu119, Glu227 and Asp151), site-3 (Ile222 and Tyr178), and site-4 (Glu276 and Glu277). Moreover presence of the 150 loop (residues 147–152), adjacent to the active site leads to the formation of closed and open conformations, enabling binding to inhibitors [26]. We have selected an open conformation structure of H1N1 complexed with zanamivir (3B7E) for our docking study. The protocol was validated by docking zanamivir in active site of monomeric unit of H1N1- NA complex. The original zanamivir pose in complex form and docked pose when superimposed showed good correlation. The docked pose of zanamivir showed interaction of its carboxylate group with Arg118 and Arg371 while the carbonyl oxygen of the carboxamide group interacted with Arg151. The guanidine amino group of zanamivir interacted with Trp178 and Glu227 whereas the glycerol hydroxyl group interacted with Glu276 and opened the new pocket by interacting with Tyr406. All above interactions were consistent with original crystal structure of zanamivir in PDB (3B7E). It was observed that in comparison with oseltamivir, zanamivir showed additional interactions provided by its guanidine and glycerol moieties, with Trp178, Glu227 and Asp151. This might be due to zanamivir’s lower susceptibility toward resistance to NA. It also highlighted that increasing molecular volume of oseltamivir, especially around the site-2, might help in more precise binding thereby decreasing the resistance of the enzyme towards the inhibitors. Furthermore, it is reported that proper molecular volume, shape and electronic charge with respect to oseltamivir is critical for activity [27]. The poses of sialic acid (endogenous ligand), zanamivir and oseltamivir in the active site of neuraminidase obtained by docking are depicted in Figure 3. Intermolecular hydrogen bonds observed between these molecules and the amino acids in the H1N1-NA active site have been tabulated in Table 1.

Figure 3: Docked pose of Sialic acid (yellow), Zanamivir (purple) and Oseltamivir (green) in H1N1-NA active site.
Various chalcone derivatives when docked in active site showed different preference of interaction towards each site based on the substituent. Hydrogen bond interactions of all chalcone derivatives are shown in Table 2.
| Amino acid residue | ChoCl | ChmCl | ChpCl | ChoH | ChmH | ChpH | ChoN | ChmN | ChpN | ChoM | ChmM | ChpM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H1N1-NA | ||||||||||||
| Arg118 | - | - | - | - | - | - | NH2…OH | - | - | - | NH2..OH | - |
| NH2..O=C | ||||||||||||
| Glu119 | - | - | - | C=O..OH | - | - | - | - | - | - | C=O..O=C | - |
| Asp151 | - | - | - | - | - | - | - | - | - | - | - | C=O..OH |
| Arg152 | - | - | - | - | NH2..O=C | - | - | NH2..O=C | - | - | - | - |
| Arg156 | - | - | - | - | - | NH2..OH | - | - | - | - | - | - |
| Trp178 | - | - | - | - | - | - | - | - | NH2..NO2 | - | - | - |
| Ser179 | C=O..OH | NH2..O=C | - | - | - | - | - | - | - | - | C=O..OCH3 | - |
| Arg192 | - | - | - | - | - | - | - | - | C=O..O=C | - | - | |
| Arg224 | - | NH2..Cl | - | - | - | NH2..O=C | - | - | - | - | - | - |
| Glu227 | C=O..OH | C=O..OH | C=O..OH | - | C=O..OH | - | C=O..OH | - | NH2..O=C | - | C=O..OCH3 | - |
| Tyr247 | - | - | - | - | - | - | - | - | C=O..OH | C=O..OH | - | - |
| Glu276 | - | - | C=O..Cl | - | C=O..OH | - | - | - | - | - | - | - |
| Glu277 | - | - | - | - | - | C=O..OH | C=O..OH | C=O..OH | - | - | - | - |
| C=O..O=C | ||||||||||||
| Arg292 | - | - | - | NH2..OH | - | - | - | NH2..NO2 | - | C=O..OH | - | C=O..O=C |
| Asn294 | - | - | NH2..Cl | - | - | - | - | NH..NO2 | - | - | - | - |
| Arg371 | - | - | - | - | - | - | NH2..NO2 | - | - | - | - | - |
| Tyr406 | - | - | - | - | - | - | C=O..NO2 | - | - | - | - | C=O..O=C |
| No. of H-Bonds | 2 | 3 | 3 | 2 | 3 | 3 | 6 | 4 | 3 | 3 | 5 | 3 |
Atoms involved in hydrogen bonding from ligand (row) to amino acid residue of H1N1-NA (column) are underlined. Last row shows number of hydrogen bond observed in case of each ligands.
Table 2: Results of docking studies of chalcone derivatives with H1N1-NA.
Based on various interactions we have selected three representative molecules for further studies. The docked poses of three selected molecules ChpM, ChpH and ChpN are shown in Figure 4. It was observed that ChpH with 4-OH and ChpN with 4-NO2 fitted predominantly into site 2, almost in a superimposable fashion except the hydrogen bond interaction of 4-OH and 4-NO2 groups in the respective molecules on ring B with Arg156. In case of ChpH, 4-OH interacted with Arg156 with one hydrogen bond while in ChpN, 4- NO2 interacted with Arg156 showing two hydrogen bonds. In both the cases hydroxyl group 2’-OH on ring A interacted to form hydrogen bonds with Glu277 and Arg224.

Figure 4: Docked pose of ChpM (yellow), ChpH (green) and ChpN (purple) in H1N1 NA active site.
In order to understand the importance of shape complementarities and electronic environment of these molecules, we have calculated vdW and electrostatic interactions (Table 3). It was observed that derivative ChpH and ChpN interacted with active site predominantly due to its electrostatic contribution especially in 150 loop. On the other hand derivative ChpM with 4- methoxy group (ring B) showed vdW interaction prominently in site 1, site 4 and in 150 loop while it was not showing electrostatic interaction in site 4 and 150 loop. This might be due to its more hydrophobic nature.
| Active site | Sialic acid | Zanamivir | Oseltamivir | ChpM | ChpH | ChpN | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| vdW | Ele | vdW | Ele | vdW | Ele | vdW | Ele | vdW | Ele | vdW | Ele | |
| Site 1 | ||||||||||||
| Arg118 | -1.91 | -0.72 | 0.4 | -12.63 | -1.79 | -11.7 | -0.59 | -6.7 | 0.3 | 5.4 | -0.45 | 3.6 |
| Arg292 | -4.44 | 2.94 | -2.93 | 6.57 | -3.59 | 4.67 | -0.43 | -13 | 0.14 | 16.41 | 0.7 | 17.7 |
| Arg371 | -1.51 | -7.07 | -1.68 | 1.27 | -1.6 | -4.99 | -0.06 | -8.15 | -0.06 | 9.66 | -0.06 | 7.98 |
| Site 2 | ||||||||||||
| Glu119 | -2.34 | 8.1 | -2.53 | -2.23 | -1.25 | 7.89 | 99.66 | 12.2 | -1.68 | -2.01 | 9.33 | 2.3 |
| Asp151 | -0.33 | -8.37 | -3.74 | -8.24 | -2.88 | -4.43 | -2.49 | -0.5 | -3.42 | 2 | -1.6 | -0.6 |
| Arg156 | -0.27 | 6.48 | -0.81 | -3.51 | -0.28 | -4.18 | -0.94 | 10.86 | -0.46 | -4.36 | 6.18 | -8.17 |
| Glu227 | -1.08 | -0.72 | -1.6 | 1.8 | -0.89 | 3.4 | -0.66 | -0.23 | -0.81 | 1.23 | -0.83 | 0.55 |
| Site 3 | ||||||||||||
| Trp178 | -1.61 | -2.01 | -1.57 | 2.3 | -1.92 | 4.15 | -0.09 | -0.4 | 10.93 | 5.1 | 16.13 | -2.1 |
| Ile222 | -1.21 | -2.9 | -1.16 | -2.12 | -1.95 | -2.12 | 1.11 | 0.05 | -2.23 | -1.8 | -2.3 | 0.13 |
| Site 4 | ||||||||||||
| Glu276 | 0.58 | -12.27 | 2.07 | -4.07 | 4.13 | -3.74 | -0.9 | 1.63 | 5.05 | -3.7 | 2 | -2 |
| Glu277 | -2.22 | 1.71 | -1.42 | 1.08 | -2.38 | 3.11 | -1.07 | 0.4 | -2.32 | -2.8 | -2.26 | -1.2 |
| 150 loop | ||||||||||||
| Val149 | -0.08 | -9.99 | -0.08 | -9.39 | -0.08 | -0.53 | -0.05 | 0.29 | -0.03 | -5 | -0.04 | -4.8 |
| Lys150 | -0.1 | -7.89 | -0.15 | -11.47 | -0.11 | -7.49 | -0.16 | 6.3 | -0.12 | -3.6 | -0.18 | 2.4 |
| Asp151 | -0.33 | -8.37 | -3.74 | -8.24 | -2.88 | -4.43 | -2.49 | -0.5 | -3.42 | 2.06 | -1.6 | -0.67 |
| Arg152 | -2.16 | -18.6 | -2.97 | -18.26 | -1.96 | -19.49 | -3.59 | 10.86 | -0.89 | -4.35 | -1.7 | -8.1 |
Table 3: Vdw interaction of chalcone derivatives with respect to amino acid residues of H1N1 NA active site.
Binding studies with MLVs
To understand the effect of ChpM, ChpH and ChpN on membrane binding, the molecules were incorporated in model membrane prepared from DPPC. The experiments were carried out using fixed concentration of ChpM, ChpH and ChpN while varying concentrations of DPPC. Results were determined from the fraction of drug bound to the MLVs. Fraction of the drug bound was determined by calculating difference in the absorbance of pure drug and when it is incorporated in different amount of MLVs. Figure 5 shows a plot of fraction of drugs bound to MLVs with increasing concentration of lipid. A plot of inverse of the fraction of drug bound vs. inverse of lipid concentration is linear. Binding constant K was calculated from the slope of the straight line. The apparent binding constants measured for different molecules were, quercetin: 1.1494 M-1, derivative ChpM: 0.8097 M-1, derivative ChpN: 0.7374 M-1, derivative ChpH: 0.6199 M-1. The extent of binding was in the order of quercetin>ChpM> ChpN >ChpH. These results were consistent with AlogP values and indicated that the molecules bound to the MLVs with variable degree of affinity which might be due to their structural differences.

Figure 5: Binding of chalcone derivatives (A) ChpM (B) ChpH (C) ChpN (D) Quercetin with increasing concentrations of lipid. Insert figure-Double inverse plot of fraction of drug bound vs. inverse of lipid concentration.
DSC studies
The effect of thermotropic aspect of drug-lipid interaction can be studied using DSC by examining the changes in the melting point (Tm) and the shape of the DSC trace. Figure 6 shows DSC curves of chalcone derivatives ChpM, ChpH, ChpN and quercetin (curves A-D). The multilamellar bilayers of plain DPPC show a pretransition at 33.74°C and a main transition at 41.34°C due to mobility of the polar choline head group and the alkyl chain respectively (Figure 6A). It was observed that the DSC curve of DPPC bilayer incorporated with derivative ChpM and ChpH (Figures 6A and 6B) at different concentrations (curves b-d), both the pre-transition peak as well as main transition shifted to lower temperature (Table 4) while the shape of endotherms remained unperturbed. The main transition peak at lower lipid concentration (1:10) shifted slightly to lower value by 1.0°C whereas at the higher concentrations (1:5 and 1:2) there was no further change in main transition temperature. The pre transition peak at lower concentration remained unchanged while at higher concentration (1:1 molar ratios) it abolished. The slight decrease in main transition temperature indicated that the molecules imparted fluidity to the bilayer in order to position themselves within the hydrophobic core. Similar behavior was observed for DPPC incorporated with quercetin (Figure 6D). However, with increasing concentration of the drug, the pretransition became broad in this case indicating interaction of the molecule with the head group as well (curves b-d). On the other hand, the DSC curve of DPPC bilayer incorporated with ChpN (Figure 6C) showed negligible change in main transition temperature, large change in pretransition temperature indicating the fluidization effect in both the hydrophilic and hydrophobic regions of the bilayer.

Figure 6: DSC plots of DPPC (50 mM) incorporated with (A) ChpM (B) ChpH (C) ChpN (D) Quercetin. The additive:lipid molar ratio are (a) 0:100 (b) 1:10 (c) 1:5 (d) 1:2.
| Drug:DPPC | ChpM | ChpH | ChpN | Quercetin | ||||
|---|---|---|---|---|---|---|---|---|
| I | II | I | II | I | II | I | II | |
| 0.0694444 | 33.4 (0) | 41.2 (0) | 33.4 (0) | 41.2 (0) | 33.4 (0) | 41.2 | 33.4 | 41.2 |
| 0 | 0 | 0 | ||||||
| 1:10 | 32.3 (1.1) | 40.2 (1.0) | 29.8 (3.6) | 40.1 (1.1) | 29.8 (3.6) | 40.9 | Broad | 40.1 |
| -0.3 | -1.1 | |||||||
| 1:05 | 31.1 (2.3) | 40 (1.2) | 29 (4.4) | 40.1 (1.1) | 29 (4.4) | 40.9 | Broad | 39.1 |
| -0.3 | -2.1 | |||||||
| 1:02 | Broad | 40 (1.2) | Broad | 40 (1.2) | Broad | 40.9 | Broad | 38.9 |
| -0.3 | -2.3 | |||||||
Bracket indicates the difference between temperatures with respect to DPPC
Table 4: DSC studies showing the pretransition (I) and maintransition (II) temperature in °C of DPPC (50 mM) with chalcone derivatives in varying molar ratio.
NMR experiments
13C NMR: To congregate more detailed information about the interactions occurring at a molecular level between lipid in model membrane and drug, i.e. chalcone derivatives (ChpM, ChpH, ChpN), 13C NMR experiments have been carried out by incorporating the compounds in the lipid bilayer. In case of ChpM and ChpN, on incorporation into the bilayer all the signals were broadened almost to baseline (Figure 7). The broadening of the signals arises due to an exchange between the bound and the free form of these drugs at intermediate time scale. Due to the broad nature of the signals it is not possible to measure both the spin lattice relaxation time (T1) and the spin–spin relaxation time (T2) which was a measure of the overall tumbling behavior and segmental motion of the molecule. In the fast tumbling range, both the T1 and T2 are large, of the order of a few seconds. These molecules lost their mobility and became strongly bound to lipid bilayer resulting in loss of motional freedom. On the other hand the 13C NMR spectrum of ChpH incorporated with lipid bilayer showed changes in drug as well as lipid signal. This further indicated that the molecule was in fast exchange between bound and free form as a result of which the drug signals remained sharp.

Figure 7: 500 MHz 13C NMR spectra of (A-C) ChpM, ChpH and ChpN respectively, incorporated into DPPC unilamellar vesicles (1:5 additive:lipid molar ratio). (D) Plain DPPC (50 mM) in D2O. All experiments are at 323 K
The results consequently indicated that binding of these molecules to DPPC was greatly dependent on the nature of the derivative. ChpH showed flexible binding characteristic, whereas ChpM and ChpN were strongly bound to the lipid bilayer and lost their motional freedom as indicated by a complete broadening of the signals.
31P NMR: The 31P NMR spectra of lipids provides a wide range of information about lipid bilayer packing, lipid head group orientation/ dynamics, and elastic properties of pure lipid bilayer. 31P NMR spectroscopy is sensitive to local motions and orientation of the phosphate group present in lipid, making it well suited for monitoring structural changes and to detect polymorphism in model membranes. The 31P NMR resonance line shape changes due to the chemical shift anisotropy (CSA) of the phosphate group coupled with the molecular motions near the head groups.
The effect of derivative ChpM, ChpH, ChpN and quercetin on the 31P line shape was measured as a function of concentration on incorporation of varying concentration of derivatives with lipid bilayers (Figure 8). In case of ChpM, 31P NMR line shape (Figures 8 (a-d)) was not affected and remained very similar to that of pure DPPC bilayer (Figure 8 (a)) and also CSA remained constant even with increasing drug concentration (Figure 9). In case of derivatives ChpH and ChpN 31P NMR line shape was not affected (Figure 8(b-d)) and remained very similar to that of pure DPPC bilayer, but CSA changed to a very large extent (Figure 9). This indicated that in all the four cases the bilayer structure remained stable. Further, the change in CSA in case of ChpH, ChpN and quercetin indicated the binding of the drug to the phosphate head group in the order ChpH> ChpN> quercetin > ChpM. ChpM showed negligible change in CSA.

Figure 8: 500 MHz 31P NMR spectra of DPPC (100 mM) multilamellar vesicles incorporated with (A) ChpM, (B) ChpH, (C) ChpN, and (D) Quercetin.

Figure 9: Concentration dependent Chemical shift anisotropy (?s=s- - s? ) for lipid incorporated with Derivatives A, B, C and D (Quercetin).
The membrane fusion is very important process in biological system. The stabilization of the lipid bilayer features as observed by DSC and NMR study further supported that the membrane fusion was restricted under these conditions. This could be a possible reason for the antiviral activity of chalcone derivatives as these molecules prevent the membrane fusion leading to their antiviral activity.
Anti-viral evaluation
Out of ten derivatives synthesised and based on docking results, we have evaluated antiviral activity of seven chalcone derivatives by in vitro cell based technique done using oseltamivir as a standard. We have used MDCK cell lines and influenza A virus strain A/Pune isolate/2009 (H1N1) for the study. It was observed that all the tested compounds had no serious effect on MDCK cells at concentrations upto 20 µM except for derivative ChpN and ChpCl which showed cytotoxic effect at concentrations at 2 µM and 0.2 µM respectively.
The virus’s HA titre is a simple number of the highest dilution factor that produced heamagglutinition [28]. The % log2 HA titre [29] reduction for each compound was calculated using Eqn 1:
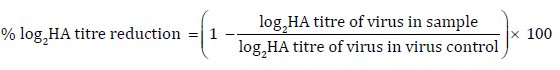
The antiviral effect of the compounds was ascertained by measuring concentration required for 50% log2 HA titre reduction (EC50) and is given in Table 5. It indicated that all compounds under the study can effectively suppress viral activity after 1 h incubation at 37°C. It was observed that standard drug oseltamivir phosphate exhibited activity at 7.1 nM. Almost all synthesized derivatives showed activity parallel to the standard. Among all derivative ChmN showed highest activity at 0.67 nM whereas lowest activity was displayed by derivative ChmM at 10.35 nM. Derivative ChpH showed activity almost equivalent to oseltamivir. All the results indicated that the chalcone derivatives showed reasonable antiviral activity.
| Code | Name | EC50 (nM) |
|---|---|---|
| A | ChpM | 3.54 |
| B | ChpH | 7.12 |
| C | ChpN | Cytotoxic |
| D | ChpCl | Cytotoxic |
| E | ChmM | 10.35 |
| F | ChmH | 8.2 |
| G | ChmN | 0.67 |
| OMVP | Oseltamivir Phosphate | 7.1 |
Table 5: Results of H1N1- HA assay of some synthesized chalcone derivatives.
Molecular docking studies indicated that various chalcone derivatives when docked in active site showed different preference of interactions towards each site based on the substituents. The membrane fusion is very important process in biological system. Antiviral drugs are known to act by inhibiting membrane fusion leading to membrane stabilizing effect. The above results of our compounds under study pointed towards stabilization of the membrane bilayer. When comparison was done between known drug oseltamivir and synthesized derivatives, it was observed that incorporation of ChpM (4-OCH3) into the bilayer, phase of multilamellar vesicles retained the bilayer phase characteristics. In case of derivatives ChpH (4-OH) and ChpN (4-NO2) though the bilayer phase remained intact, there was a small change in motional rigidity and very little perturbation to bilayer was observed. These observations were further supported by a recent DSC study. In case of DSC, result indicated that the bilayer remained intact showing little perturbation which changed motional rigidity of the vesicle imparting slight membrane fluidity. On the basis of these results we conclude that chalcone derivatives synthesized by us have promising activity against H1N1 virus. All the compounds under study showed different mode of binding in the active cavity and almost all showed good antiviral activity. The most promising ChpM and ChmN can be explored further by evaluating its interaction with neuraminidase using in vitro enzyme assay protocols. Also, the candidate molecules were tested on seasonal influenza virus which further propels the activity to be tested on pandemic and mutant strains of the influenza virus.
M. A. Kanyalkar thanks Indian Council of Medical Research (ICMR), New Delhi for funding computational facilities at Prin. K. M. Kundnani College of Pharmacy through Adhoc research scheme (58/27/2007-BMS). A. S. Chintakrindi also thanks ICMR, New Delhi for Junior Research Fellowship (58/27/2007-BMS). The authors gratefully acknowledge the National facility for High Field NMR located at TIFR for providing NMR and DSC facilities and National Institute of Virology, Pune, India for H1N1 virus.