Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Editorial - (2008) Volume 1, Issue 5
Transcriptomic and proteomic technologies are vogues in analyzing biological entities. The integrality of their probe sets or searching databases is the prerequisite of full identification, which could be estimated by their coverages over genome. After the completion of human genome atlas with painstaking effort, expression profiling technology becomes routine analysis in both transcriptome and proteome levels. The mainstream strategies of such profiling, based on expression microarrays and mass spectrum, are enclosed in probe set and sequence database respectively. It is conceivable that their integralities, i.e. their coverages to the whole human genome, determine their capacity in cataloguing transcriptome and proteome.
After the completion of human genome atlas with painstaking effort, expression profiling technology becomes routine analysis in both transcriptome and proteome levels. The mainstream strategies of such profiling, based on expression microarrays and mass spectrum, are enclosed in probe set and sequence database respectively. It is conceivable that their integralities, i.e. their coverages to the whole human genome, determine their capacity in cataloguing transcriptome and proteome.
For the estimate of their integralities, human genome repository in NCBI known as Entrez Gene was used as genome background. Totally 36,545 items including 25,611 Protein Coding Genes (PCGs), and 1,114 RNA Coding Genes (RCGs) beyond PCG were compiled in version 2007- 05-21.
In Microarray-based Transcriptome Profiling (MbTP), two most widely used human “whole genome” expression microarray, Affymetrix HG-U133 plus 2.0 and Agilent Human Genome Whole-Four-Plex 44K were chosen as representatives. The HG-U133 plus 2.0 contains 54,675 probe sets which can be mapped to 21,083 genes in Entrez Gene, and the 41,000 probe sets in the Whole-Four-Plex covers 19,610 genes. For PCGs, both of them have close but only medium coverage (76.23% in the HG-U133 plus 2.0, and 71.31% in the Whole-Four-Plex). Their specific parts include 1,899 and 638 PCGs respectively. Their combination can just slightly promote the PCG coverage up to 78.72%. The 5,450 PCGs lost by MbTP could be categorized as “hypothetical” (2,291 genes) or “similar to”/“-like” (2,012 genes), and “others” including 234 ORF-related, 75 zinc finger proteins, 36 immunity products and 802 other functional-known genes. It is notable that 233 members of olfactory receptor gene family and 49 keratin coding genes were lost by MbTP. Obviously, current MbTPs could not fully identify the proteome-encoding transcriptome. On the other hand, for RCGs, the MbTP covers only less than 15% coding genes of miscellaneous RNA (miscRNA), small nucleolar RNA (snoRNA), and small nuclear RNA (snRNA); even worse, none of the two microarrays have any ability to detect rRNA and tRNA (see Table 1). Evidently, those non-protein-coding RNAs should be explored only by virtue of other specialized microarrays.
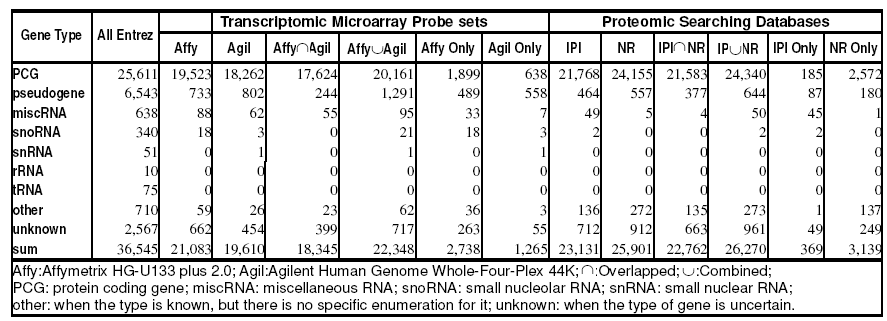
Table 1: Coverage of most widely used transcriptomic probe sets and proteomic searching databases to human genome.

Figure 1: Flowchart of MbTP and MSbPP datasets construction.
The latest Affymetrix “whole genome” expression microarray Human Gene 1.0 ST covers 19,915 genes with 18,570 PCGs. Those numbers are even less than those of the HG-U133 plus 2.0. The replacement of the Human Gene 1.0 ST to the HG-U133 plus 2.0 will result in the loss of other 2,920 genes including 2,038 PCGs. So we finally choose the later as the “whole genome” representative from Affymetrix series. Honestly, addition of the Human Gene 1.0 ST to the MbTP can slightly raise the total gene coverage (61.15%->ú64.60%) and PCG coverage(78.72%->ú81.48%). Of the additional 708 PCGs, 533 have known functions. Moreover, all the lost 233 olfactory receptor genes and partial keratin genes (31 of 49 lost) have been retrieved in the Human Gene 1.0 ST. Even so, the HG-U133 plus 2.0 is still with more but not too much preponderance coverages of all the genes and PCGs.
Probe set design should keep up with genome databases, but is fettered by period of products and somewhat lag behind genome updating. As a remission, some extensional probe sets which corresponding to low quality annotated sequences in current genome database can be designed in microarray in advance. Besides of the “enclosed” transcriptome detection technique, Serial Analysis of Gene Expression (SAGE) and further developed Massively Parallel Signature Sequencing (MPSS) are two major complementary techniques to transcriptome microarray. They can detect transcripts from unknown (novel) genes.
In Mass Spectrum-based Proteome Profiling (MSbPP), protein sequence databases can follow more tightly with updating of genomic sequences. The International Protein Index (IPI) in EBI and the “Non-Redundant” protein database (NR) in NCBI are two dominant databases for mass spectrum data searching. IPI updates monthly and NR updates in real time through web interface. IPI Human v.3.32 released in 2007-08-06 contains 67,524 items covers 21,768 PCGs; NR-Human from FTP download (release 2007- 08-08) and web download (release 2007-08-10) contains 175,947 and 410,546 items respectively, totally covering 24,155 PCGs. The both proteomic databases reach significantly higher PCG coverage (IPI 84.99%, NR 94.31%), especially the later. IPI-mapped PCGs are almost included in those of NR, only with 185 specifically mapped PCGs (see Table 1). We can construct an MSbPP searching database by combining IPI and NR. It will achieve a little bit higher PCG coverage at 95.04%. Even so, there are also 1,271 PCGs lost by MSbPP, including 482 hypothetical proteins, 269 “similar to”/“-like” type proteins, 165 ORFs related, 55 zinc finger proteins, 29 immunity products and 268 genes with other functions. Evidently, although MSbPP searching databases are much better in integrality than the probe sets of transcriptome profiling, but remain insufficient for full identification of proteome.
When we talk about the completion of transcriptome and proteome, two kinds of “completion” should be differentiated: Other than genome blue print, transcriptome is with temporal (expression status) variable; proteome is with both spatial (subcellular) and temporal variables. The completion of all infinite statuses is of course infeasible. If the“completion” refers to grasp all elements (mRNAs or proteins) in a measurement, such as low abundance issues, we think that is possible to achieve the goal by technique improving.