Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2014) Volume 2, Issue 6
We analysed the clinical and hematological features in 41 patients of seven families, including 21 ET patients with a proven MPLS505N mutation and 20 relatives with thrombocythemia reported in the medical records. Out of the 41 MPLS505N mutated individuals 15 major thrombotic episodes in 14 members (34%) were reported as Budd-Chiari syndrome age 17 in 1, deep vein thrombosis leg age 41 in 1,ecclampsia and fetal in 1, stroke at ages 43, 72, 76 and 80 in 4 and myocardial infarction at ages between 31-81 years, median 52 years. Fourteen out of 21 well documented MPLS505N mutated ET patients had no splenomegaly and were free of major thrombosis during follow-up at ages between 2 and 76 years (mean 31 years). Eight MPLS505N mutated patients had myelofibrosis (MF) from grade MF1 in 5 to grade MF2 in 3 at ages between 28-80 years (mean 48 years), which was associated with mild to moderate splenomegaly (spleen length diameter 14.5 to 18 cm). Six anemic cases at hemoglobin levels between 10 and 11.9 g/dL had platelet counts between 317 and 963 × 109 /L. Among 15 family members 9 died from thrombosis in 3, hypocellular myelofibrosis (two of them at age 76 and 80 years) in 3, and cancer or undefined in 3 cases. The maximum life expectancy of MPLS505N family members with thrombocythemia was 50% at 80 years, and 90% at 80 years of non-affected family members without thrombocythemia. The clinical presentation in 30 ET patients with acquired MPL505N mutation (9 males and 21 females, age 22-84 (mean 56 years of whom 18 had the W515L and 12 the W515K) was featured by a high incidence of major arterial event in 23%, venous thrombosis in 10%, aspirin responsive microvessel disturbances in 60%, and major hemorrhage in 7%. The only abnormal laboratory finding in MPL mutated ET was increased platelet counts, 956+331 × 109 /L in all and slight splenomegaly in 5 (17%). Bone marrow histology from patients ET carrying the MPL505N mutation consistently displayed a normocellular bone marrow with clustered small and large to giant megakaryocytes with hyperlobulated stag-horn nuclei and no features of polycythemia vera (PV) in blood and bone marrow.
Keywords: Myeloproliferative Neoplasm; Essential Thrombocythemia; Myelofibrosis; Congenital or acquired MPL mutations.
In the 1990s, studies on murine leukemia and oncogenes led to the recognition of a new member of the hematopoietin receptor super family [1], which was discovered as the product of the gene c-mpl, the normal cellular homologue of the oncogene v-cmpl, the transforming principle of a murine myeloproliferative leukemia virus, responsible for a panmyloid transformation [2]. This was followed by the molecular cloning and characterisation of Mpl, the human homologue of the c-mpl and v-mpl [3-5]. The new receptor was than rapidly recognized as being the thrombopoietin receptor (TpoR) by the demonstration that antisense oligonucleotides of c-mpl inhibited the colony-forming of megakaryocyte progenitors by Wendling and Vainchenker [4]. The Mpl ligand became the key to the identification of TPO, which was cloned in 1994 by five independent groups [6-11]. The MPL ligand is identical to TpoR and labeled as megakaryocyte growth and development factor (MDGF) [10-13]. Human TPO has all the functions ascribed to MDGF, and all MDGF-like activity can be neutralized by soluble recombinant Mp [1].
Thrombopoietine (TPO) is encoded by the human TPO gene and located on chromosome 3q271. The mature TPO molecule is composed of 32 amino acids. The TPO Receptor (TpoR) is encoded by the myeloproliferative leukemia (MPL) gene, which is located on chromosome 1p34. TPO and its ligand MPL are involved in nearly every step of megakaryocyte development including mobilisation of hematopoietic stem cells, stimulating megakaryocyte proliferation and lineage differentiation. TPO deficient mice show a marked decrease in the number of megakaryocytes and platelets, accompanied by significant reduction of all lineage hematopoietic progenitor cells [14-16]. These animals are the counterpart of congenital recessive amegakaryocyte thrombocythemia (CAMT), a pedriatric disease resulting from the loss of function of the MPL gene in newborns with recessive severe thrombocytopenia who later develop severa anemia in early childhood. TpoR or MPL exists as a transmembrane receoptor in megakaryocytes and platelets, that has no intrinsic kinase activity, but associates with the cytoplasmic tyrokinase JAK2. Upon TPO ligand binding the TpoR/MPL receptor undergoes a conformational shift followed by cross-activation of two JAK2 molecules. TPO stimulation results in phophorysation of MPL-bound JAKs and the subsequent activation of several down-stream pathways.
Megakaryopoiesis Plasma TPO, TPOR/CMPL Receptor and Platelet Production
TPO messenger studies showed that the main site of TPO production is in the liver and kidneys, an expression pattern similar to EPO [1]. It is known that hepatoblastoma and nephroblastoma may be accompanied by thrombocytosis due to increased TPO production and increased plasma TPO levels. During megakaryopoiesis the hematopoietic stem cell pass through a stage that they are still bipotent and have the capacity to become either erythroid cells or megakaryocytes under the influence of EPO and TPO respectively. The megakaryoblasts undergo a number of endomitotic reduplications and mainly megakaryocytes with 16-32N chromosomes and only a few 64-128N (hyperploidization) chromosomes are formed [1]. The last stage I the formation of filamentous extrusions called proplatelets, which fragment into platelets. TPO levels in serum are about three to four times higher than in EDTA plasma, because TPO is stored in platelets in high concentration and released during coagulation.
Plasma TPO levels in various blood and coagulation disorders reveal an inverse relationship between amegakaryocytic thrombocytopenia due to loss of function mutation of cMPL (Figure 1) [1]. In patients with pancytopenia and thrombocytopenia caused by acute myleloid leukemia (AML) or myelodysplastic syndrome (MDS) TPO levels are high (Figure 2) very likely due megakaryocytopenia in the bone marrow [1].
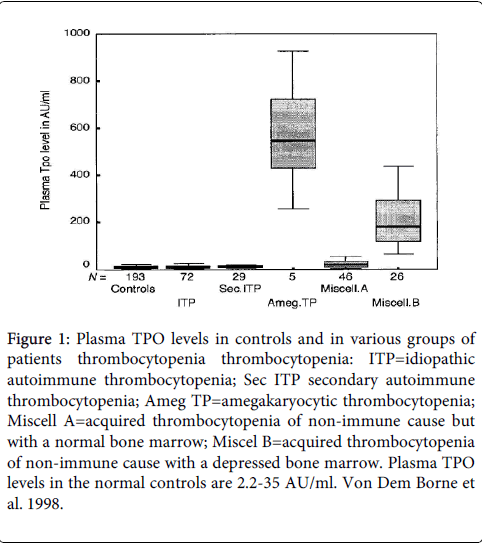
Figure 1: Plasma TPO levels in controls and in various groups of patients thrombocytopenia thrombocytopenia: ITP=idiopathic autoimmune thrombocytopenia; Sec ITP secondary autoimmune thrombocytopenia; Ameg TP=amegakaryocytic thrombocytopenia; Miscell A=acquired thrombocytopenia of non-immune cause but with a normal bone marrow; Miscel B=acquired thrombocytopenia of non-immune cause with a depressed bone marrow. Plasma TPO levels in the normal controls are 2.2-35 AU/ml. Von Dem Borne et al. 1998.
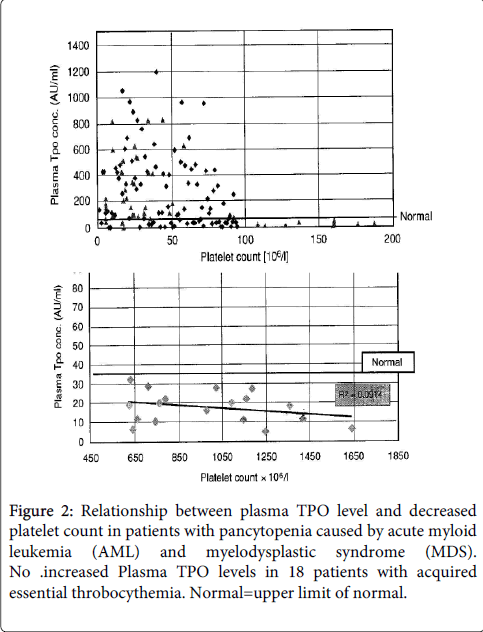
Figure 2: Relationship between plasma TPO level and decreased platelet count in patients with pancytopenia caused by acute myloid leukemia (AML) and myelodysplastic syndrome (MDS). No .increased Plasma TPO levels in 18 patients with acquired essential throbocythemia. Normal=upper limit of normal.
Plasma TPO levels are normal in immune thrombocytopenia due to an increased platelet destruction (Figure 2) and in patients with increased platelet count in reactive thrombocytosis and in essential thrombocythemia. This contrasts with low EPO levels concentrations found in JAK2V617F mutated ET, prodromal PV and classical PV.
Plasma TPO levels in ET (JAK2V617F positive and JAK2 wild type) are not down regulated. This difference reflects the fact that EPO is regulated at the level of production, while TPO is regulated largely by binding to platelets and their rate of peripheral consumption. In most clinical studies serum is used thus not only free plasma TPO, Serum TPO measures the total of plasma TPO plus TPO released from platelets during blood coagulation.
In ET this may lead to the measurement of high serum TPO concentrations in ET. Von den Borne found that plasma TPO levels in essential thrombocythemia are not increased (Figure 2) [1].
Congenital Het Caused by the MPLSER505ASN Mutation
Congenital gain of function mutation in the TPO gene on chromosome 3q27 results in increased levels of plasma TPO levels, which induce a physiological activation of the TPO?TPOR/MPL signalling pathway in three reported families with hereditary essential thrombocythemia [17-20]. This results in hyperproliferation of large mature megakaryocytes and platelet count complicated by platelet-mediated microvascular complications. In a previous report we reviewed the presenting features and the natural history of two families with hereditary ET (HET) and secondary myelofibrosis caused by a gain of function mutation in the TPO gene [21]. In 2004 Ding et al. described the first case of congenital ET in the pedigree of a Japanese family caused by a G to A nucleotide substitution at position 1073 in exon 10 of the MPL gene leading to the exchange os serine for asparaginase at position 505 (MPLS505N) [22]. Ding et al. demonstrated that the presence of the germ line MPLS505N mutation is associated with autonomous activation in the MPL downstream signaling pathways, both in vitro in cells transfected with the mutant and in vivo in platelets obtained from affected individuals. The authors clearly demonstrated that cells expressing MPLS505N showed autonomous phosphorylation of both Mek1/2 and Stat5 down signaling transduction pathways but the clinical course of the disease in terms of vascular complications were not reported in family members carrying the MPLS505N mutation.
Teofili et al. described in 2010 eight Italian families positive for the MPLS505N mutation and reported on the clinical manifestations and hematological features in 41 patients of seven families, including 21 ET patients with a proven MPLS505N mutation and 20 relatives with thrombocytosis [23]. The family history of 41 individuals reported 15 major thrombotic episodes in 14 members (34%): Budd-Chiari syndrome age 17 in 1, deep vein thrombosis leg age 41 in 1,ecclampsia and fetal in 1, stroke at ages 43,72, 76 and 80 in 4 and myocardial infarction at ages between 31-81 years, median 52 years. These patients were not on aspirin at time of the occurrence of major thrombotic event and after a major thrombotic event they were treated with hydroxyurea or pegylated interferon (IFN) according to Italian guidelines (high thrombotic risk ELN) [24]. The overall survival and thrombosis-free survival in 41 affected family members with hereditary ET with MPLS505N thrombocythemia (Family members with thrombocytosis, Figure 3) was compromized as compared to normal overall and thrombosis-free survival in non-affected family members without thrombocythemia (Family members without thrombocytosis, Figure 4). Clinical manifestation of aspirin responsive microvascular disturbances including erythromelalgia, migraine-like atypical cerebral transient ischemic attacks (MIAs) and visual ischemic disturbances usually precede the occurrence of major thrombosis in ET and PV when not on aspirin. The indication of aspirin in 15 out of 21 MPLS505N mutated ET cases was headache not otherwise specified, and none experienced bleeding complications during follow-up. Fourteen out of 21 documented MPLS505N carriers were free of major thrombosis during follow-up at ages between 2 and 76 years (2, 4, 7, 20, 25, 28, 31, 34, 42, 69 and 76 years). There was no splenomegaly in 13 these 14 cases without a history of thrombosis.
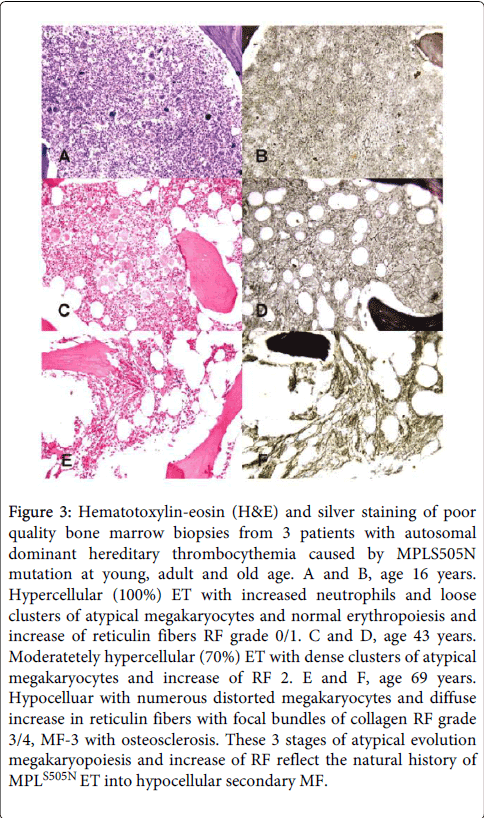
Figure 3: Hematotoxylin-eosin (H&E) and silver staining of poor quality bone marrow biopsies from 3 patients with autosomal dominant hereditary thrombocythemia caused by MPLS505N mutation at young, adult and old age. A and B, age 16 years. Hypercellular (100%) ET with increased neutrophils and loose clusters of atypical megakaryocytes and normal erythropoiesis and increase of reticulin fibers RF grade 0/1. C and D, age 43 years. Moderatetely hypercellular (70%) ET with dense clusters of atypical megakaryocytes and increase of RF 2. E and F, age 69 years. Hypocelluar with numerous distorted megakaryocytes and diffuse increase in reticulin fibers with focal bundles of collagen RF grade 3/4, MF-3 with osteosclerosis. These 3 stages of atypical evolution megakaryopoiesis and increase of RF reflect the natural history of MPLS505N ET into hypocellular secondary MF.
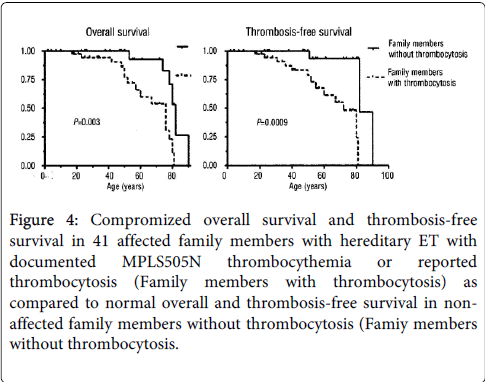
Figure 4: Compromized overall survival and thrombosis-free survival in 41 affected family members with hereditary ET with documented MPLS505N thrombocythemia or reported thrombocytosis (Family members with thrombocytosis) as compared to normal overall and thrombosis-free survival in nonaffected family members without thrombocytosis (Famiy members without thrombocytosis.
Three stages of atypical evolution megakaryopoiesis and increase of RF in bone marrow biopsies reflecting the natural history of MPLS505N ET from normocelluar or hypercellular ET into hypocellular secondary MF is shown in Figure 3.
At age 16 years a hypercellular (100%) ET with increased neutrophils and loose clusters of atypical megakaryocytes ws associated with and normal erythropoiesis and increase of reticulin fibers RF grade 0/1.
At age 43 years moderate hypercellularity (70%) in ET and dense clusters of atypical megakaryocytes was associated with an increase of reticuin fibers grade 2 (RF 2=MF 1).
At age 69 years a hypocelluar bone marrow with numerous distorted megakaryocytes was associated with diffuse increase in reticulin fibers (RF grade 3 to 4) and focal bundles of collagen myelofibrosis MF-3 with osteosclerosis.
In this cohort of 21 MPLS505N patients four cases presented with major thrombosis (19%); stroke at age 76 and 80 in 2, DVT/TIA at age 41/43 in 1 and myocardial infraction at age 31 in 1. Eight MPLS505N patients had myelofibrosis (MF) [25] grade MF1 in 5 and grade MF2 in 3 at ages 76, 55, 80, 28, 23, 33, 67, which was associated with mild to moderate splenomegaly (spleen length diameter 14.5 to 18 cm). Five of these MF patients were treated for several years with hydroxyurea (HU) in 3, interferon (IFN) in 1 and HU/IF in 1, and four of these five MF cases had anemia as a side effect of hydroxyurea and/or myelofibrotic transformation. All MF1 or MF2 patients had normal leukocyte counts counts except one female at age 72 years. Six anemic cases at hemoglobin levels between 10 and 11.9 g/dL had platelet counts between 317 and 963 × 109/L. At hemoglobin levels above 12 g/dl platelet counts ranged from 605 to 1726 × 109/L. Leukocyte counts were completely normal except leucocytosis above 10 in 3 young affected children (age 4 to 7 years) and one female at age 72. Among 15 family members 9 died prematurely of major thrombosis in 3, anemia with hypocellular myelofibrosis (2 cases at age 76 and 80 years, Figure 3), and liver cirrhosis, gastric cancer or undefined in 1 case each. Comparing all family members without thrombocythemia the overall survival and thrombosis-free survival was significantly shortened in MPLS505N mutated thrombocythemia patients. The maximum life expectancy of MPLS505N family members with thrombocythemia was 50% at 80 years, and 90% at 80 years of non-affetected family members without thrombocythemia (Figure 4). This analysis strongly suggest that the loss of life expectancy is mainly due to major thrombosis and myelofibrotic transformation since 2 of 3 MPLS505N cases died from hypocellular myelofibrosis died at old ages of 76 and 80 years.
Acquired MPL515 mutated Essential Thrombocythemmia
The JAK2 kinase activity in MPN is not only dependent on the amount of heterozygous and homozygous JAK2V617F mutant protein, but may also be influenced by the various steps upstream or downstream the signalling pathways including MPL, JAK2, STAT-3. This has been demonstrated in animal models overexpressing c-MPL [8,9]. MPL transgenic mice manifested with typical features of ET with a fourfold increase of platelet count, increased colony formation of megakaryocytes, and increase of clustered enlarged megakaryocytes in the bone marrow. The ET animals appeared healthy, had a very slight decrease of hematocrit (0.39 versus 0.42 in controls) despite an increase of bone marrow EEC, and survived normally with no evidence of myelofibrosis in the bone marrow [8,9]. The first case of congenital ET by Ding et al. in 2004 in a Japanese family caused by the germline MPLS505N mutation [22] and the discovery of the JAK2V617F mutation by Vainchenker et al. in 200526 as the cause of ET and PV has led to the discovery of the MPLW515L and MPLW515K.somatic mutations as the cause of acquired ET and secondary myelofibrosis [26-29]. Within the JAK2 wild type MPN, there is a small subgroup who carry an acquired gain of function mutation of the MPL receptor as the cause of ET: 3% in the Vannucchi study [29], and 8.5% in the UK studies [30,31]. In contrast to JAK2V617F mutated trilinear MPN, patients with JAK2 wild type PT carrying the MPL515 mutation have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis, do not evolve into overt PV during follow-up, have normal serum EPO and ferritin levels, and show pronounced megakaryoctic proliferation of small and large (giant) megakarocytes and no increase of erythropoiesis in the bone marrow [29].
Vannucchi et al. studied [30] Essential Thrombocthemia (ET) patients carrying the MPL515 mutation, 9 males and 21 females, age 22-84 (mean 56) years [29]. The clinical presentation at diagnosis and follow-up was remarkable with a high incidence of major arterial event, 23%, venous thrombosis, 10%, microvessel disturbances, 60%, and major hemorrhage, 7%. The only abnormal laboratory finding was increased platelet counts, 956+331 × 109/L together with hemoglobin values in the lower range of normal (13.4+1.3 g/l), normal white blood cells (8.8+3.1 × 109/L), slight increase of LDH (459+182 U/L) and splenomegaly in only 5 (17%) of 30 MPL515 mutated ET cases of whom 18 had the W515L and 12 the W515K allele mutated. Mutation allele burden was greater than 50% in half of MPLW515K patients compared to 17% of MPLW515L mutated ET patients. MPL515 and JAK2V617F mutations coexisted in 3 with MPLW515L and in 5 with MPLW515K allele mutations. General features of bone marrow reports revealed significantly reduced erythropoiesis and decreased cellularity in MPLW515L/K patients, associated with increased number of clustered small and large megakaryocytes, but no significant increase in reticulin fibrosis (RF). Activation of MPL by thrombopoietin enhances normal platelet function and abnormal activation of ET platelets by thrombopoietin preincubation has been described by Akkerman et al. [32]. Consequently, Vannucchi et al. hypothesized that platelets from MPLW515L/K mutated ET patients present consitutively enhanced reactivity (hypersensitive) of the mutated platelets to explain the high incidence of aspirin responsive microvascular disturbances and major arterial thrombotic events in acquired MPL mutated ET [29]. The Dutch family with hereditary ET due to a gain of function mutation in the TPO gene had life-long increased plasma TPO levels and presented at young and adult age recurrent erythromelalgia complicated by acrocyanosis of a few toes followed by gangrene and amputation of toe, which typically responded to low dose aspirin but not by Coumadin [21] thereby preventing the occurrence of major thrombotic during lifelong follow-up.
Bone Marrow Histology in Acquired MPL515 Mutated ET and MF
In 2008 we studied bone marrow histopathology in 12 cases with JAK2 wild type ET carrying the MPL515 mutation kindly provided by the courtesy of Dr. Vannucchi, Florence, Italy. Bone marrow histology from patients with JAK2 wild type ET carrying the MPL515 mutation consistently displayed clusters small and large megakaryocytes with a greater number of giant megakaryocytes with hyperlobulated stag-horn nuclei in a normal cellular bone marrow and no increase of erythropoiesis. Bone marrow histology of two representative cases of MPL515 mutated ET are shown Figures 5 and 6.
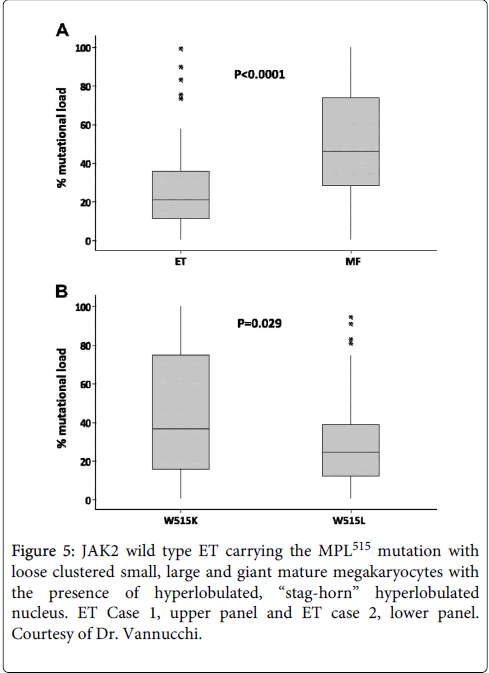
Figure 5: JAK2 wild type ET carrying the MPL515 mutation with loose clustered small, large and giant mature megakaryocytes with the presence of hyperlobulated, “stag-horn” hyperlobulated nucleus. ET Case 1, upper panel and ET case 2, lower panel. Courtesy of Dr. Vannucchi.
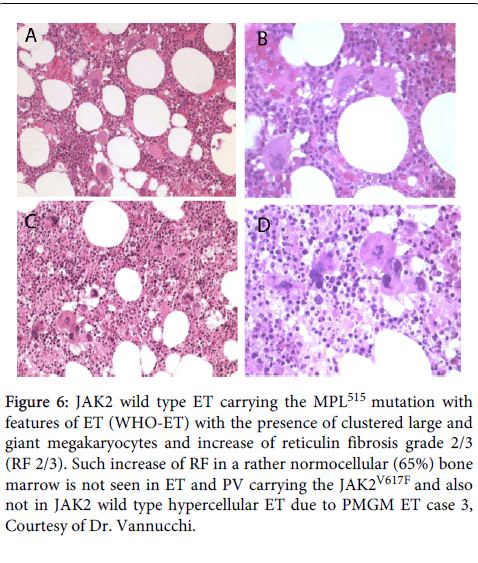
Figure 6: JAK2 wild type ET carrying the MPL515 mutation with features of ET (WHO-ET) with the presence of clustered large and giant megakaryocytes and increase of reticulin fibrosis grade 2/3 (RF 2/3). Such increase of RF in a rather normocellular (65%) bone marrow is not seen in ET and PV carrying the JAK2V617F and also not in JAK2 wild type hypercellular ET due to PMGM ET case 3, Courtesy of Dr. Vannucchi.
As compared to bone marrow histopathology in JAK2V617F mutated ET (Figure 7) there were significant differences on three points. First, the megakaryocytes in MPL515 mutated PT are larger than in ET and PV (Figures 5 and 6), whereas the megakaryocytes in JAK2V617F mutated ET are not larger than in PV and show similar pleomorphic megakaryocytes morphology as in PV (Figure 7).
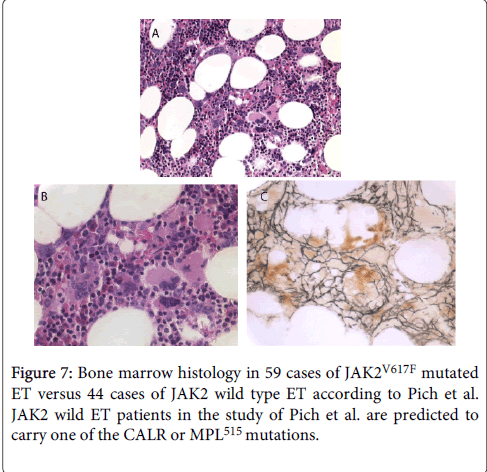
Figure 7: Bone marrow histology in 59 cases of JAK2V617F mutated ET versus 44 cases of JAK2 wild type ET according to Pich et al. JAK2 wild ET patients in the study of Pich et al. are predicted to carry one of the CALR or MPL515 mutations.
Second, there was local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryoctyes in JAK2V617F mutated ET (Figure 7), but not in JAK2 wild type PT carrying the MPL515 mutation.
Whether such differences of megakaryocyte morphology in bone marrow biopsies are characteristic enough to distinguish normocellular ET with low JAK2 mutation load from JAK2 wild type ET carrying the MPL515 mutation respectively by expert hematopathologists remains to be evaluated in prospective clinical and basic research studies.
Case Report on the Natural History of MPL515 Mutated ET
We studied the natural history of a 41-year old woman born in 1995 who presented in January 1996 with MPLW515L mutated ET and a one year history of tingling prickling sensations in fingers and hand, vertigo and attacks of frontal headaches. Laboratory features at time of diagnosis were, hemoglobin 12.8 g/L, hematocrit 0.39, leukocytes 7.5 × 109/L, normal LDH and spleen size on echogram 12.6 cm (normal value <12 cm). Platelet counts were 790 × 109/L with highest platelet count of 1996 × 109/L in October 1996. Molecular biology analysis were negative for the JAK2V617F, CALR and ASXL1 mutations. Bone marrow histology showed a normal cellularity of about 40%, no increase of erythropoiesis, and prominent increase of large to giant megakaryocytes with hyperlobulated nuclei even stag-horn forms with fine chromatin (Figure 8). Sporadically, small megakaryocytes with less lobulated nuclei were present. Fine perivascular reticulin fibers (RF grade 1) consistent with prefibrotic myelofibrosis MF 0. Treatment consisted of low dose aspirin for the relief of microvascular disturbances, pegylated interferon (IFN) was not tolerated and subsequent treatment consisted of hydroxyurea from 1996 to 1999 was followed by anagrelide from January 1999 untill she developed refractory pancytopenic anemia and thrombocytopenia in 2004. Bone marrow histology in 2004 showed a hypocellular bone marrow with dysmorphic small to large megakaryocytes and only slight increase of reticulin fibers RF grade 1 to 2 (Figure 9). Hematopioetic stem cell transplantation (HSCT) in February 2005 of a matched (10/10) unrelated donor and non-myeloablative conditioning (Busulfan-Cytarabine-ATG) was followed by rapid engraftment and complete hematological remission. She is more than 9 years alive and well in 2014 and beyond.
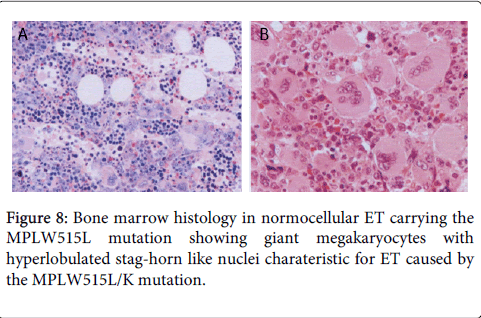
Figure 8: Bone marrow histology in normocellular ET carrying the MPLW515L mutation showing giant megakaryocytes with hyperlobulated stag-horn like nuclei charateristic for ET caused by the MPLW515L/K mutation.
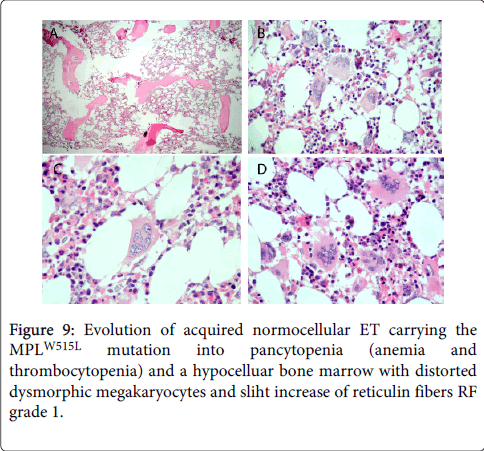
Figure 9: Evolution of acquired normocellular ET carrying the MPLW515L mutation into pancytopenia (anemia and thrombocytopenia) and a hypocelluar bone marrow with distorted dysmorphic megakaryocytes and sliht increase of reticulin fibers RF grade 1.
In a European collaborative study, Jones et al. determined the MPL mutation load in 138 W515K/L MPN cases: ET, n=99; MF, n=36; unclassified MPN, n=331. The overall median W515K/L mutation levels in ET was 21%, which were significantly lower than in MF patients, 46% (P<.001, Figure 9). Twenty nine MPL515 homozygous cases (mutation load more than 50% in 17 W515L and in 12 W515K MPN cases) had a diagnosis of ET in 12, MF in 15 and accelerated/transformed MPN in 2. The overall mutation levels in 106 MPLW515L cases were lower, 25%, as compared to 37% in 32 MPLW515K cases (P=0.02), thereby confirming the observations of Schnittger et al. in 35 cases with four different MPL515 mutations in ET or MF34. In this German cohort of 324 JAK2 wild type ET, any MPLW515 mutation was detected in 19 patients (5.9%), and of 104 JAK2 wild type MF, 10 MPLW515 mutations (9.6%) were detected. In addition two novel MPL mutations W515R and W515K were found. We request in 2013 and 2014 the German MPN investigators Schnittger, Reiter and Kvasnicka to collect BM biopsies for expert evaluation of the WHO defined clinical molecular and pathological (WHO-CMP) [33-36] features in their cohort of 35 MPL mutated ET and MF patients. Beer et al. described bone marrow biopsies at diagnosis of 13 patients with MPL mutations: 2 S505N, 2 W515K and 9 W515L MPN patients [30]. As compared to JAK2V617F positive ET and JAK2/MPL wild type ET, the bone marrow biopsies from the MPL mutant group were less cellular (P<0.001 and P<0.003 with age). Both elytroid and granulocytic cellularity were reduced in the MPL mutant group. (P<0.001) indicating the absence of PV features. Overall, bone marrow histology in MPL mutant patients revealed more isolated megakaryocytic proliferation at diagnosis with a reduction of erythroid and overall cellularity compared to JAK2V617F positive and JAK2/MPL wild type ET (Figure 10) [37]. Serum EPO levels in MPL mutation patients were significantly higher than in JAK2V617F positive ET (prodromal PV) [35,36]. In contrast to JAK2V617F positive ET. Beer et al. found no endogenous erythroid colonies (EEC) in any of 5 evaluated MPLW515L cases (4 ET and 1 MF). At the basic research level Chaligne et al. demonstrated that the two MPL mutations W515L and W515K induced a spontaneous megakaryocyte growth in culture with an overall normal response to thrombopoietin (TPO), but the erythroid progenitors remained EPO dependent and did not show spontaneous erythroid colony (EEC) formation (Figure 10) [21,33].
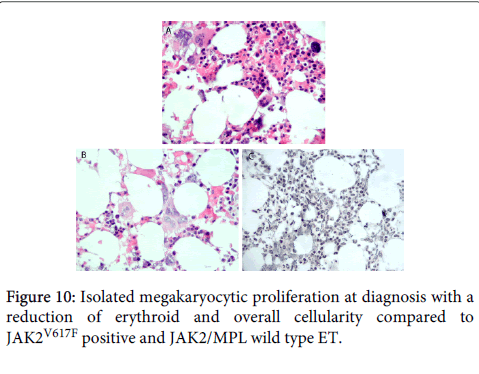
Figure 10: Isolated megakaryocytic proliferation at diagnosis with a reduction of erythroid and overall cellularity compared to JAK2V617F positive and JAK2/MPL wild type ET.