Journal of Cancer Science and Research
Open Access
ISSN: 2576-1447
ISSN: 2576-1447
Short Communication - (2018) Volume 0, Issue 0
Keywords: Immune checkpoint blockade; Colorectal cancer; PDI
Immunotherapy, working through immune checkpoint blockade has achieved notable responses in multiple tumor types including malignant melanoma, renal cell carcinoma, non-small cell lung cancer, bladder carcinoma, Hodgkin’s lymphoma, triple-negative breast carcinoma as well as head and neck cancer [1]. However, CRC appears to be one of the tumor types that shows a poor response to immune checkpoint inhibitors, apart from the MSI CRC subtype which accounts for about 5% [2] of advanced and metastatic CRC [3]. So why don’t immune checkpoint inhibitors work in MSS CRC?
In this brief review we will consider the following:
a) Results of clinical trials of colorectal cancer with immune checkpoint inhibitors
b) Mechanisms underlying immune escape by CRC through immunoediting, which is often caused by down regulating MHC class I and class II; down regulation of antigen processing TAP enzymes; decreased expression of co-stimulatory molecules; infiltration of the tumor by regulatory T cells.
c) Hypothesis that histone deacetylase (HDAC) inhibitors reverse immunoediting.
Results of clinical trials with immune checkpoint inhibitors
Two phase I clinical trials of anti PD-1 (BMS936558, Nivolumab) and anti PD-L1 (BMS936559) antibodies were carried out in a variety of cancer types including colorectal cancer (19 colorectal cancer out of 296 cancer patients and 18 colorectal cancer out of 207 cancer patients respectively). Notably, only 1 patient from the metastatic-CRC cohort in the BMS936558 clinical trial, who had a PD-L1 positive tumor, showed a complete response after 6 months’ treatment of BMS936558 and had no signs of tumor recurrence after 3 years. This patient’s tumor was also mismatch repair deficient [4-6].
Since somatic mutations have the potential to encode “non-self ” immunogenic antigens, it was hypothesized those tumors with mismatch-repair deficiency that can lead to thousands of somatic mutations may be responsive to immune checkpoint inhibitors. 41 patients with or without mismatch repair deficient advanced cancer were recruited into a phase II clinical trial of Pembrolizumab conducted by Le et al. Patients were treated with Pembrolizumab intravenously 10 mg/kg every 2 weeks. They were separated into 3 cohorts of MMR-deficient colorectal cancer, MMR-proficient colorectal cancer and MMR-deficient non-colorectal cancer. The primary end point for the first two cohorts was the immune-related objective response rate and the immune-related progression-free survival rate at 20 weeks. The primary endpoint for the third cohort was the immune-related progression-free survival rate at 20 weeks.
The results showed that the 2 groups with MMR-deficient colorectal or non-colorectal cancer had the higher rate of immune-related objective response (40% and 71%) and immune-related progressionfree survival at 20 weeks (78% and 67%) compared to the MMRproficient colorectal cancer group (0% and 11%).
Interestingly, 1782 versus 73 somatic mutations per tumor in mismatch repair-deficient tumors and mismatch repair-proficient tumors was demonstrated (P=0.007), and higher somatic mutation loads were associated with prolonged progression-free survival (P=0.02). Rash/pruritus, pancreatitis, and thyroiditis/hypothyroidism were found to be the most common treatment-related adverse events, occurring in approximately 10% of patients. This phase II clinical trial proved that patients with mismatch-repair deficient tumors associated with a heavy load of somatic mutations respond to anti-PD1 therapy [3]. Based on this promising study, phase III trials investigating the effect of anti-PD-1 therapy in MSI-H colorectal cancer have been initiated.
Mechanisms of cancer immune evasion in colorectal cancer
Cancer immunoediting is the term used to describe the dual role of the immune system in host- protection and tumor-sculpting. It consists of 3 phases (also known as the 3Es): elimination, equilibrium, and escape [7,8].
In the elimination phase, congenital and adaptive immune cells recognize and destroy the accumulating tumor cells before the clinical manifestations occur. When the tumor cells break through the elimination phase and proceed into the equilibrium state, immunologic mechanisms begin to work to prevent tumor outgrowth [9,10]. Tumors escape due to the ever growing population and the changes in their response to immunoselective pressures and/or to increased tumor-induced immunosuppression or immune system deterioration. [8,11] There are multiple possible mechanisms causing immunoediting: 1) Down regulation of HLA-I and 2. 2) Down regulation of antigen processing TAP enzymes. 3) Decreased expression of co-immunostimulatory molecules; and 4) Infiltration of the tumor by regulatory T cells.
Down regulation of human leukocyte antigen class I: According to Menon et al. over 70% of colorectal cancers undergo a downregulation of human leukocyte antigen class I, the so called human MHC [12]. Thus, tumor cells with downregulated but not completely depleted HLA-1 expression can avoid T cell and NK cell-mediated immune surveillance, and may be to some extent correlated with poor prognosis [13]
Also, a study with large sample numbers (462 tumors) reported that down regulation of MHC-I is an independent marker for poor prognosis in early stage colorectal cancer. This implies that if the immune response does occur in early stage disease, it may eliminate micrometastases which have an intact antigen presenting system.
Down regulation of antigen processing tap enzyme: Crucial for the process of translocating peptide from cytoplasm to the endoplasmic reticulum(ER), the transporter associated with antigen processing (TAP) system is another factor influencing immunoediting. TAP transporters load peptide fragments from tumor cellular antigens onto major histocompatibility complex (MHC) I molecules. Loaded MHC-I leave the ER and display the antigen on the cell surface, permitting their recognition by CD8+ T lymphocytes, which can induce a cellular immune response and cell destruction. [14].
Ras oncogenic transformation, found in approximately 40% of CRC, is associated with reduced TAP and proteasome subunit low molecule protein (LMP) mRNA expression. This results in incomplete peptide transport and peptide loading of MHC class I molecules, resulting in reduced stability of expression of the MHC class I complex on the cell surface. [15] Down regulation of or depleted TAP1 has been found in different tumor types with frequencies ranging from 10 to 84%. [16-20] Interestingly, interferon-gamma [15] and interferon- alpha [21] treatment can enhance expression of TAP, low molecular protein (LMP) and MHC class I molecules in parental and ras transformed fibroblasts.
Kasajima et al. explored the in vivo association of TAP and MHC class I antigen and their impact on prognosis in colorectal cancer. Immunohistochemical assessment of TAP1, TAP2 and MHC class I antigen expression in 336 sporadic colorectal carcinomas was performed in this study (Figure 1). They found TAP1 and TAP2 expression to be significantly associated with MHC class I antigen expression (P<0.001). Increased density of CD8 (+) TIL was predominantly found in TAP1, TAP2 and MHC class I antigenpositive cases, and tumors with CD8+ lymphocytic infiltration had an improved prognosis [22].
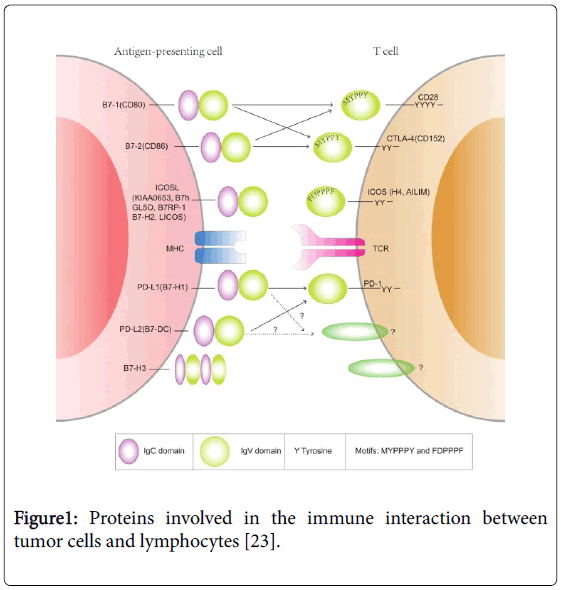
Figure 1: Proteins involved in the immune interaction between tumor cells and lymphocytes [23].
Decreased expression of co-stimulatory molecules: The interaction between co-stimulatory molecules expressed on the cell surface of antigen presenting and tumor cells and the receptors on immune cells activate a range of intracellular signals to activate T lymphocytes [24,25]. Common co-stimulatory molecules include B7-1/B7-2: CD28/ CTLA-4 family and TNF: TNFR family. Different pairs of costimulatory molecules have different ways to interact with each other, for example, one receptor may be activated with one type of ligand while restricted by other type of ligand or one receptor may be linked to two or more ligands [26-28]. Most tumors lack or downregulate the expression of positive costimulatory molecules such as B7-1 (CD80) and B7-2 (CD86) [29-33].
Infiltration of the tumor by t regulatory cells: Regulatory T cells act to down regulate effector immune responses. They have a distinct phenotype with the expression of CD4, CD25 and FoxP3. Most studies have concluded that high levels of infiltrating regulatory T cells are correlated with poor prognosis in many kinds of tumors including colorectal cancer [34-38]. However, some studies showed that high density infiltration of T reg cells are correlated with an improved outcome in treating cancer [39] which may be explained by differences in methodological sub classification of T reg cells. Both murine models and human in vitro models show that depletion of T reg cells induces immune responses against tumor-associated antigens, so it is mechanistically plausible that regulatory T cells are correlated with poor prognosis in colorectal cancer [40-43].
Mismatch repair deficiency (Microsatellite instability)
Deficient mismatch repair (dMMR or microsatellite instability, MSI) is one of the key genetic mechanisms driving the occurrence and progression of CRC. There are several genes controlling DNA mismatch repair function including MSH2, MLH1. One consequence of dMMR is that these tumor cells carry a very high neo antigen load due to the high frequency of mutations, increasing the likelihood of immune recognition. Perhaps unsurprisingly, microsatellite unstable colon tumors appear have a strong lymphocyte infiltration [44] and have a significantly better prognosis than MSS colorectal cancer, especially in stage II disease [45].
Tumor lymphocytic infiltration
Takemoto showed that stroma-infiltrating lymphocytes (SIL) were found in approximately the same number in high grade microsatellite instability (MSI-H) patients (20%) and low grade microsatellite instability (MSI-L) or MSS tumors (12.8%). However, significant differences of intra-tumor cell-infiltrating lymphocytes (ITCIL) were shown between MSI-H CRC and MSI-L or MSS CRC patients (41.7% vs . 4.3%, respectively (P<0.001)). Furthermore, the prognosis of the tumors with higher ITCIL counts was better than the less infiltrated ones. [46] In addition, increased PD-L1 expression has been found at the invasive edge of MSI-H tumors.
All the characteristics mentioned above, as well as the recent definition of highly immunogenic neo-antigens expressed in MSI-H tumor cells, suggest that MSI-H CRCs induce a protective host immune response that may reduce the incidence of metastasis formation and which might explain the better outcome in this patient group [47-49].
Can we reverse immunoediting by treating with histone deacetylase (hdac) inhibitors
Researchers have hypothesized that strategies which increase expression of T-cell chemokines and T-cell infiltration of tumors would be capable of enhancing response to PD-1 blockade. There is evidence to suggest that histone deacetylase inhibitors (HDACi) [50,51] are capable of inducing expression of these chemokines in tumor and increasing immune recognition.
It has been reported that increased histone acetylation induced by HDAC inhibitors results in the increase expression of MHC molecules and other molecules involved in antigen processing and presentation [52-55]. Also it can increase expression of tumor antigens recognized by cytotoxic T lymphocytes (CTLs) and ligands for NK activating receptors [56,57]. The HDACi romidepsin, induced a strong antitumor response against KRAS mutant NSCLC tumors in mice which correlated with T cell infiltration of the tumor, and CD8 T-cell infiltration in human lung tumors has been shown to increase after HDACi vorinostat treatment [58,59]. The combination of the HDAC I depsipeptide and very low concentrations of the cytotoxic antimetabolite 5-fluorouracil (5-FU) induces apoptosis synergistically and up regulates MHC class II in human colon cancer HCT-116 cells [60].
Based on these and many other preclinical study results, several clinical trials have been initiated to evaluate whether the combination of HDAC inhibitors and anti-PD1 therapies can improve tumor responses by enhancing the CD8 T cell infiltration. (NCT02638090, NCT02437136, NCT02697630, NCT01928576, NCT024353620 and NCT02708680) These trials may give an indication if HDAC inhibitors can improve response to anti-PD1 agents in the coming future.
As mentioned in this article, it is the loss of function of cellular immune system, that complies possible reason causing no response to checkpoint blockade therapy in colorectal cancer. During the cell deterioration process of CRC, the deficiency of immuno stimulatory signal presentation and the activation of immunological checkpoints may suppress the immunosurveillance [61]. One clinical approach to targeting the mechanisms that underlie immune evasion in colorectal cancer may be to combine immune checkpoint and HDAC inhibitors to restore immunoreactivity and enhance tumor cell kill.