Transcriptomics: Open Access
Open Access
ISSN: 2329-8936
ISSN: 2329-8936
Research Article - (2013) Volume 1, Issue 1
Background: Transposable Elements (TEs) have long been regarded as selfish or junk DNA having little or no role in the regulation or functioning of the human genome. However, over the past several years this view came to be challenged as several studies provided anecdotal as well as global evidence for the contribution of TEs to the regulatory and coding needs of human genes. In this study, we explored the incorporation and epigenetic regulation of coding sequences donated by TEs using gene expression and other ancillary genomics data from two human hematopoietic cell-lines: GM12878 (a lymphoblastoid cell line) and K562 (a Chronic Myelogenous Leukemia cell line). In each cell line, we found several thousand instances of TEs donating coding sequences to human genes. We compared the transcriptome assembly of the RNA sequencing (RNA-Seq) reads with and without the aid of a reference transcriptome and found that the percentage of genes that incorporate TEs in their coding sequences is significantly greater than that obtained from the reference transcriptome assemblies using Refseq and Gencode gene models. We also used histone modifications chromatin immunoprecipitation sequencing (ChIP-Seq) data, Cap Analysis of Gene Expression (CAGE) data and DNAseI Hypersensitivity Site (DHS) data to demonstrate the epigenetic regulation of the TE derived coding sequences. Our results suggest that TEs form a significantly higher percentage of coding sequences than represented in gene annotation databases and these TE derived sequences are epigenetically regulated in accordance with their expression in the two cell types.
<Keywords: Gene expression; Transcription; Transposable elements; Exonization; Epigenetics; RNA-seq; DNAseI HS; Alu; LTR Retrotransposons
RNA: Ribonucleic acid; RNA-Seq: RNA sequencing; TE: Transposable Element; ChIP-Seq: Chromatin Immunoprecipitation Sequencing; DNA: Deoxyribonucleic Acid; DHS: DNAseI Hypersensitivity Site; ENCODE: Encyclopedia of DNA Elements; LTR: Long Terminal Repeats; IAP: Intracisternal A Particle; TSS: Transcription Start Site; NCBI: National Center for Biotechnology Information; GEO: Gene Expression Omnibus; RIKEN: RikagakuKenkyujo; UCSC: University of California-Santa Cruz; FDR: False Discovery Rate.
A substantial fraction of the eukaryotic genome is comprised of transposable elements (TEs). In the human genome, more than half of the euchromatic sequence can be attributed to TEs. Despite their profusion, TEs have historically been regarded as selfish genomic parasites or junk DNA that have little to no contribution in the functioning of the human genome [1,2]. The aforementioned studies argued that TEs can maintain and proliferate in the host genome solely because of their ability to out-compete the host genome in terms of their replication ability, and are therefore not constrained to have a functional contribution necessary for selection. These studies shaped the paradigm for research in the field by discouraging the search for the functional relevance of TE sequences in the human genome.
Over the last several years, scientists discovered gene features such as promoters, UTR’s and exons derived from TE insertions that regulate transcription [3,4]. Exonization is a process whereby genes acquire new exons from non-protein-coding DNA sequences, often times TEs due to the presence of internal splice site-like structures within their sequences [5]. A classic example is the agouti locus in mice, where its expression results in a yellow coat color, obesity, diabetes, and tumor susceptibility [6]. The transcription of the agouti gene is influenced by a cryptic promoter originating from an intracisternal particle (IAP) element, a member of the LTR-Retrotransposon family, which generates a transcript that reads through the agouti gene. The number of transcripts originating from the IAP element depends on its methylation state where a de-methylated IAP element is capable of generating transcripts resulting in an increase in the expression of the agouti gene.
Indeed there are several instances where TEs have been implicated in functionally contributing to the human genome [7]. These contributions include TEs providing regulatory and coding sequences such as transcription factor binding sites, promoters, exons, and enhancers [8-12]. As is the case with the Agouti locus described above, TE derived gene features are often epigenetically regulated to cater to the functional needs of the cell type they are in. We have recently shown that TEs donate several hundred canonical promoters and transcription start sites to the human genome that are epigenetically modified in such a way as to facilitate cell type specific expression [13,14]. Similarly, we have also demonstrated that TEs donate thousands of unique enhancers that are modified by epigenetic histone modifications and are functionally relevant in driving cell type specific gene expression [14,15].
These studied provided the motivation to explore the global contribution of TE derived gene features in various human cell types and their potential role in gene regulation. The availability of RNA sequencing (RNA-Seq) and epigenetic data from various human cell lines in the Encyclopedia of DNA Elements (ENCODE) project provides an unprecedented survey of the landscape of transcription genome-wide and, in a limited way, catalogues transcripts initiated from repetitive elements in a cell line specific manner [16]. However, there is a paucity of information regarding epigenetic regulation of TEderived transcripts. To that end, we used the RNA-seq data as well as the epigenetic data provided by the ENCODE project in two human hematopoietic cell lines (GM12878 and K562)in order to ascertain the interplay between the exonization of TEs into coding sequences and epigenetic regulation of transcription on a genome-wide scale. GM12878 is a lymphoblastoid cell line derived from a female donor of northern and western European descent whereas K562 is a cancer cell line from a female patient suffering from Chronic Myelogenous Leukemia (CML). In each cell line, we found several thousand instances of TEs donating coding sequences to human genes and that TE exonization is significantly greater in the reference guided transcriptome assembly (Refseq [17] and Gencode [18]) than an assembly without the aid of a reference transcriptome. In addition, integration of histone modifications chromatin immunoprecipitation sequencing (ChIP-Seq) data, cap analysis of gene expression (CAGE) data and DNAseI hypersensitivity site (DHS) data allowed us to postulate that TEs contribute a substantial fraction of human coding sequences that are epigenetically regulated in accordance with the gene expression in the two cell types.
Datasets
RNA-seq data from GM12878 and K562 cell lines generated by Caltech were downloaded from the University of California Santa Cruz (UCSC) genome browser’s ENCODE downloads section and is also available in the NCBI GEO repository under accession #GSE23316. In each cell line, two replicate libraries of (75bpx2) paired-end reads were used for the analysis.
ChIP-seq data in GM12878 and K562 cell lines for four histone modifications (H3K4me3, H3K9ac, H3K27ac, H3K36me3) commonly found at transcription start sites and exons, were obtained as aligned tag files generated by the Broad institute and downloaded from the ENCODE section of the UCSC genome browser [19,20]. The data is also available in the NCBI GEO repository under accession #GSE29611.
Open chromatin data generated by Duke University under the ENCODE project using DHS mapping were also obtained as ‘Peaks’ file from the UCSC genome browser and is also available in the NCBI GEO repository under accession #GSE32970.
CAGE data generated by the RIKEN group for ENCODE cell lines, were also obtained from the UCSC genome browser as “CAGE Clusters” of loci deemed significantly enriched for CAGE tags against a background tag distribution. The raw data is also available in the NCBI GEO repository under accession #GSE34448.
The aforementioned datasets were co-located with transposable element (TE) annotation using custom Perl scripts as described by Repeat Masker version 3.2.7. The criteria for delineating TE derived RNA-seq exons were taken as any overlap between an exon and a TE with respect to genomic coordinates. Similarly, all other datasets (Histone modifications, DHS, CAGE) were co-located using genomic overlap between the TE annotation and the respective tag/peak/cluster densities.
RNA-seq analysis
Raw paired-end sequence reads of length 75bpx2 with an insert size of 200bp were mapped individually to the reference human genome (hg19) using Bowtie (version 0.12.7) allowing at most 2 mismatches and multiple alignments for a read pair [21]. The TopHat (Cahnge oveer all in article) program was used with default parameters to ascertain splice junctions in the mapped reads [22]. Cufflinks (version 1.0.3) was used to assemble transcripts and estimate their abundance given a reference transcriptome specified by the Refseq and Gencode (version 7) gene models [18,23,24]. As shown in (Figure S1), we used Bowtie to map the RNA-Seq reads to the hg19 genome, and then used TopHat to detect splice junctions and Cufflinks to assemble the transcriptome using two different techniques in order to determine the extent of TEs incorporation. First we built the transcriptome using reference annotations from Refseq [17] and Gencode [18] to ‘guide’ the assembly of RNA-seq reads. We also built the transcriptome without the aid of a reference gene model to yield a ‘transcript-reference-free’ characterization of RNA-seq reads. We co-located TE-annotations with RNA-seq data resulting from each assembly process and discovered that the fraction of TE-derived transcripts and exons is several folds higher in non-reference guided assembly. We further evaluated these data for functional signatures such as epigenetic histone modifications present at promoters and exons, cap analysis of gene expression (CAGE) data which identifies transcript start sites (TSS), and DNAseI hypersensitive sites (DHSs) which locate actively transcribing regions as characterized by open chromatin.
Statistical analyses
A Student’s t-test-was used to compare gene expression transcripts from the non-reference guided assembly between GM12878 and K562 cell lines. False discovery rate (FDR) adjusted p-values (q-values) were used to control for multiple testing and to determine significance at q-value<0. 05. Gene Ontology and functional enrichment of differentially expressed genes were performed using the DAVID Bioinformatics Resources v6. 7 [25,26]. Pearson’s correlation (r) was also used to ascertain the relationship between various trends. The significance of a trend was determined by approximating the sampling distribution of r to the Student’s t-distribution using the formula:
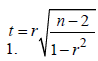
TE-derived human gene transcripts
We obtained RNA-seq data generated by Caltech for the ENCODE project in GM12878 and K562 cell lines. These data were processed using the Bowtie/TopHat/Cufflinks pipeline as described in the Materials and Methods section. The reads were first mapped to the reference human genome (hg19) using Bowtie and the TopHat program was used to map splice junctions [21,22]. The third and final step in this pipeline is quantification by Cufflinks which assembles the transcripts and estimates their abundance [23,24,27]. Normalized transcript abundance is used as a measure of expression of the respective genes. The assembly and annotation of RNA-seq transcripts by Cufflinks can be aided by providing a reference transcriptome such as Refseq to assign the mapped reads and splice junctions to known transcripts. This process generally results in a higher number of transcripts discovered but is biased towards the transcripts that exist in the reference annotation. Cufflinks can also build the transcriptome without the use of reference annotation which can be used to detect a greater number of novel transcripts that do not exist in gene model databases.
We ran Cufflinks in three different ways: without the aid of a reference transcriptome, using Refseq as reference annotation, and using Gencode as reference. This process resulted in vastly different results as illustrated in Figure 1 and Figure 2. In both cell lines, the use of a reference transcriptome during transcript assembly by Cufflinks resulted in the discovery of a considerably larger number of transcripts and exons. Transcriptome assembly using Refseq yielded the highest number of transcripts and exons, with (GM12878:1.87M and K562:1.92M) exons, and (GM12878:326K and K562:330K) transcripts. This was followed by a Gencode guided assembly which detected (GM12878:1.11M and K562:1.11M) exons and (GM12878:195K and K562:195K) transcripts in the two cell lines. On the other hand, nonreference guided assembly was able to discover only (GM12878:325K and K562:305K) exons and (GM12878:70K and K562:60K) transcripts in the two cell lines. Compared to non-reference guided assembly, these figures represent approximately 3X and 6X increase in the number of transcripts and exons discovered by Gencode and Refseq guided assembly, respectively (Figure 3 and Table 1).

Figure 1: Comparison of Gencode guided versus non-reference guided assembly of transcripts and exons.The larger circles represent all transcripts and exons whereas the smaller circles represent TE-derived transcripts and exons (a) GM12878 exons (b) GM12878 transcripts (c) K562 exons (d) K562 transcripts.

Figure 2: Comparison of Refseq guided vs non-reference guided assembly of transcripts and exons.The larger circles represent all transcripts and exons whereas the smaller circles represent TE-derived transcripts and exons (a) GM12878 exons (b) GM12878 transcripts (c) K562 exons (d) K562 transcripts.

Figure 3: TE-derived (a) transcripts and (b) exons Illustrated as a fraction of total.
| Transcripts | Exons | |||
| GM12878 | K562 | GM12878 | K562 | |
| Refseq | 19.6 | 13.4 | 4.2 | 3.7 |
| Gencode | 12.1 | 12.1 | 4.6 | 4.7 |
| Non-reference | 43.5 | 39.3 | 11.2 | 9.8 |
Table 1: Percentage of TE-derived transcripts and exons as a function of total transcripts and exons in GM12878 and K562 cell lines, refer to Figure 3.
To ascertain the contribution of transposable elements to human gene coding sequences, we co-located the transcripts and exons from each of the three assemblies with TE annotation as provided by Repeat Masker [28]. The term exonization in the context of TEs is described as the process by which a TE sequence is adapted to serve as an exon by the host genome. A transcript or exon is considered ‘TE-derived’ if any part of its coding sequence overlaps with TE annotation. The fraction of TE-derived coding sequences varies widely between the three transcriptome assemblies. As such, the non-guided assembly yields the highest proportion of TE-derived genes compared to Gencode and Refseq guided assemblies (Figure 3). These results indicate that TEderived human gene coding sequences are vastly under-represented in the reference transcriptome annotations.
Having recognized the substantial under-representation in the fraction of TE-derived exons, we sought to determine the number of these coding sequences that are common between the reference and non-reference guided assemblies. To that end, we found all transcripts or exons from different builds that have common start and end positions in the genome. In order to compensate for possible imprecision in the prediction of transcripts and exon coordinates, we allowed a 10bp wiggle room in the comparison of start and end sites of transcripts and exons from different transcriptome assemblies. We found that a very small number of TE-derived transcripts and exons are in fact common between the reference and non-reference guided assemblies (Figures 1 and 2). Since the total number of transcripts and exons discovered by the non-reference guided assembly is considerably smaller than that of reference guided assembly, it follows that a proportionally small number of TE-derived exons are in fact represented in this dataset. In other words, the total number of TEs-derived exons detected by the non-reference guided assembly may only represent a small percentage of the actual number of TEs involved presumably in serving the coding needs of the human genome. As such, these results offer an unbiased estimate of the percentage of human coding sequences derived from TEs and indicate that the contribution in absolute terms may be significantly higher. Thus, these results reveal a significantly higher level of TE contribution to human exons than previously thought.
TE-derived first exons
We evaluated the contribution of TEs in donating various gene segments including TSS (transcription start site) and first exons and compared it to subsequent exons. We report here that TEs are significantly over-represented in donating TSS and first exons as opposed to later exons (Figure 4 and Table 2). In the GM 12878 cell line (36,464/325,351) or 11.2% of all exons are derived from TEs, whereas (30,778/70,669) or 43.5% of first exons appear to be derived from TEs. Similarly, in the K562 cell line (30,126/305,792) or 9.9% of all exons are donated by TEs while (23,951/60,895) or 39.3% of first exons are TEderived. Furthermore, we found 1291 transcripts (supplemental Table S1) mapped to 69 genes (Table 3) which are differentially expressed (q-value<0.05) between the GM12878 and K562 cell lines and have TEs inserted into the first exon of the genes. These genes primarily have molecular functions in the immunoglobulin receptor family class and enrich for the Gene Ontology immune response biological process (FDR<0.1). These data seem to support the model illustrated by the agouti gene in mouse where an IAP element (LTR Retrotransposon) residing upstream of the agouti locus generates a transcript that reads through the agouti gene [29]. In the process, the IAP element donates a transcription start site and is primed to serve as the first exon for the agouti gene. Our data suggest that this model is a common route through which TEs donate an alternative TSS and the first exon that can affect the transcription of nearby genes.

Figure 4: Fraction of TE-derived exons in all exons vs first exons. TE-derived exons form a significantly higher proportion of first exons compared to all exons (a) larger circle represents all exons while the smaller circle represents first exons and (b) TE-derived exons expressed as a percentage of total exons.
| All exons | First exons | ||
|---|---|---|---|
| GM12878 | TE derived | 11.2 | 43.5 |
| Non TE derived | 88.8 | 56.5 | |
| K562 | TE derived | 9.9 | 39.3 |
| Non TE derived | 90.1 | 60.7 |
Table 2: Percentage of TE-derived first exons as a fraction of all exons in GM12878 and K562 cell lines, refer to Figure 4.
| Representative transcript | Uni Gene Cluster | Gene name | Gene symbol |
|---|---|---|---|
| NM_003975 | Hs.103527 | SH2 domain containing 2A | SH2D2A |
| NM_001039477 | Hs.10649 | Chromosome 1 open reading frame 38 | C1orf38 |
| NM_001146310 | Hs.107101 | Chromosome 1 open reading frame 86 | C1orf86 |
| NM_019089 | Hs.118727 | Hairy and enhancer of split 2 (Drosophila) | HES2 |
| NM_018420 | Hs.125482 | Solute carrier family 22, member 15 | SLC22A15 |
| NM_033312 | Hs.127411 | CDC14 cell division cycle 14 homolog A (S. cerevisiae) | CDC14A |
| NM_001765 | Hs.132448 | CD1c molecule | CD1C |
| NM_016074 | Hs.13880 | BolA homolog 1 (E. coli) | BOLA1 |
| NM_001080471 | Hs.142003 | Platelet endothelial aggregation receptor 1 | PEAR1 |
| NM_001042747 | Hs.1422 | Gardner-Rasheed feline sarcoma viral (v-fgr) oncogene homolog | FGR |
| NM_001766 | Hs.1799 | CD1d molecule | CD1D |
| NM_004672 | Hs.194694 | Mitogen-activated protein kinase kinasekinase 6 | MAP3K6 |
| NM_148902 | Hs.212680 | Tumor necrosis factor receptor superfamily, member 18 | TNFRSF18 |
| NM_030812 | Hs.2149 | Actin-like 8 | ACTL8 |
| NM_003528 | Hs.2178 | Histone cluster 2, H2be | HIST2H2BE |
| NM_001166294 | Hs.22587 | Synovial sarcoma, X breakpoint 2 interacting protein | SSX2IP |
| NM_014215 | Hs.248138 | Insulin receptor-related receptor | INSRR |
| NM_173452 | Hs.333383 | Ficolin (collagen/fibrinogen domain containing) 3 (Hakata antigen) | FCN3 |
| NM_006824 | Hs.346868 | EBNA1 binding protein 2 | EBNA1BP2 |
| NM_001007794 | Hs.363572 | Choline/ethanolamine phosphotransferase 1 | CEPT1 |
| NM_147192 | Hs.375623 | Diencephalon/mesencephalon homeobox 1 | DMBX1 |
| NM_002529 | Hs.406293 | Neurotrophic tyrosine kinase, receptor, type 1 | NTRK1 |
| NM_003517 | Hs.408067 | Histone cluster 2, H2ac | HIST2H2AC |
| NM_001193300 | Hs.408846 | Sema domain, immunoglobulin domain (Ig), transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 4A | SEMA4A |
| NM_052941 | Hs.409925 | Guanylate binding protein 4 | GBP4 |
| NM_015696 | Hs.43728 | Glutathione peroxidase 7 | GPX7 |
| NM_001143778 | Hs.437379 | ArfGAP with SH3 domain, ankyrin repeat and PH domain 3 | ASAP3 |
| NM_030764 | Hs.437393 | Fc receptor-like 2 | FCRL2 |
| NM_001033082 | Hs.437922 | V-mycmyelocytomatosis viral oncogene homolog 1, lung carcinoma derived (avian) | MYCL1 |
| NM_198715 | Hs.445000 | Prostaglandin E receptor 3 (subtype EP3) | PTGER3 |
| NM_003196 | Hs.446354 | Transcription elongation factor A (SII), 3 | TCEA3 |
| NM_005101 | Hs.458485 | ISG15 ubiquitin-like modifier | ISG15 |
| NM_001195740 | Hs.462033 | Chromosome 1 open reading frame 93 | C1orf93 |
| NM_001040195 | Hs.464438 | Angiotensin II receptor-associated protein | AGTRAP |
| NM_001048195 | Hs.469723 | Regulator of chromosome condensation 1 | RCC1 |
| NM_024772 | Hs.471243 | Zinc finger, MYM-type 1 | ZMYM1 |
| NM_002959 | Hs.485195 | Sortilin 1 | SORT1 |
| NM_001199772 | Hs.485246 | Proteasome (prosome, macropain) subunit, alpha type, 5 | PSMA5 |
| NM_178454 | Hs.485606 | DNA-damage regulated autophagy modulator 2 | DRAM2 |
| NM_002744 | Hs.496255 | Protein kinase C, zeta | PRKCZ |
| NM_001143989 | Hs.511849 | Neuroblastoma breakpoint family, member 4 | NBPF4 |
| NM_003820 | Hs.512898 | Tumor necrosis factor receptor superfamily, member 14 (herpesvirus entry mediator) | TNFRSF14 |
| NM_004000 | Hs.514840 | Chitinase 3-like 2 | CHI3L2 |
| NM_025008 | Hs.516243 | ADAMTS-like 4 | ADAMTSL4 |
| NM_001178062 | Hs.516316 | Sema domain, transmembrane domain (TM), and cytoplasmic domain, (semaphorin) 6C | SEMA6C |
| NM_021181 | Hs.517265 | SLAM family member 7 | SLAMF7 |
| NM_022873 | Hs.523847 | Interferon, alpha-inducible protein 6 | IFI6 |
| NM_152493 | Hs.524248 | Zinc finger protein 362 | ZNF362 |
| NM_000760 | Hs.524517 | Colony stimulating factor 3 receptor (granulocyte) | CSF3R |
| NM_001135585 | Hs.530003 | Solute carrier family 2 (facilitated glucose/fructose transporter), member 5 | SLC2A5 |
| NM_033504 | Hs.534521 | Transmembrane protein 54 | TMEM54 |
| NM_024901 | Hs.557850 | DENN/MADD domain containing 2D | DENND2D |
| NM_005529 | Hs.562227 | Heparansulfate proteoglycan 2 | HSPG2 |
| NM_001408 | Hs.57652 | Cadherin, EGF LAG seven-pass G-type receptor 2 (flamingo homolog, Drosophila) | CELSR2 |
| NM_033467 | Hs.591453 | Membrane metallo-endopeptidase-like 1 | MMEL1 |
| NM_024980 | Hs.632367 | G protein-coupled receptor 157 | GPR157 |
| NM_002143 | Hs.632391 | Hippocalcin | HPCA |
| NM_014787 | Hs.647643 | DnaJ (Hsp40) homolog, subfamily C, member 6 | DNAJC6 |
| NM_052938 | Hs.656112 | Fc receptor-like 1 | FCRL1 |
| NM_152890 | Hs.659516 | Collagen, type XXIV, alpha 1 | COL24A1 |
| NM_175065 | Hs.664173 | Histone cluster 2, H2ab | HIST2H2AB |
| NM_207397 | Hs.664836 | CD164 sialomucin-like 2 | CD164L2 |
| NM_001114748 | Hs.668654 | Chromosome 1 open reading frame 70 | C1orf70 |
| NM_144701 | Hs.677426 | Interleukin 23 receptor | IL23R |
| NM_001013693 | Hs.710255 | Low density lipoprotein receptor class A domain containing 2 | LDLRAD2 |
| NM_001037675 | Hs.714127 | Neuroblastoma breakpoint family, member 1 | NBPF1 |
| NM_152498 | Hs.729552 | WD repeat domain 65 | WDR65 |
| NM_001127714 | Hs.729693 | Human immunodeficiency virus type I enhancer binding protein 3 | HIVEP3 |
| NM_001164722 | Hs.77542 | Platelet-activating factor receptor | PTAFR |
Table 3: Significantly differentially expressed genes (q-value<0.05) between GM12878 and K562 cell lines and have TE inserted into the first exon of the genes.
Age and relative contribution of TE families
All major families of TEs are represented in contributing transcripts in the two cell types studied. We evaluated the relative contribution of various TE families in donating coding sequences in the RNA-seq transcripts by normalizing the number of transcripts attributed to a TE family with the background genomic abundance of the family. Different families of TEs have significantly (χ2test p-value<0. 001) different contributions in providing coding sequences as illustrated in Figure 5.

Figure 5: Relative contribution of TE-families in donating coding sequence to human gene transcripts. Absolute counts normalized by genomic background abundance for each family.
We determined the relative age of each TE family by using their average divergence (millidiv) from a consensus ancestral sequence. Using this approach, we classified the TE families in the following order according to age, from youngest to the oldest: Alu, L1, LTRRetrotransposons, DNA-transposons, L2 and MIR. We observed that the relative contribution of each TE family increases with increasing age of the family with the exception of Alu elements. Intuitively, older TE families have had a longer period of time for individual elements to be adopted by the genome and thus have relatively higher contribution compared to the younger families. Our observations are consistent with this phenomenon as older TE families are significantly more enriched in gene features. On the other hand, the youngest family of TEs i.e., Alu elements do not follow this paradigm and are highly enriched in donating coding sequences. It has previously been shown that Alu elements are enriched in gene rich regions which can be attributed to their over-representation in donating coding sequences [30].
Epigenetic regulation of TE-derived genes
To strengthen the functional relevance of TE-derived exons, we evaluated the various epigenetic markers that are known to mark active exons. To that end, we downloaded histone modification data for four histone modifications (H3K4me3, H3K9ac, H3K27ac, and H3K36me3), DHS data as well as CAGE data. These four histone modifications have been shown to mark actively transcribing TSSs and exons [31-33]. Similarly, CAGE data is an established technique to determine the TSSs for actively transcribing genes [34]. Finally, DHSs are used to identify open chromatin and are shown to be correlated with activation of gene expression [35,36].
These data were mapped to the TE-derived exons using their genomic loci. We divided the TE-derived exons into 100 bins according to their expression in the two cell lines and plotted their average epigenetic modification against their expression (Figures 6 and 7). In both cell lines, the expression of TE-derived exons is strongly positively correlated with the histone modifications, DHSs, and CAGE clusters. These epigenetic indicators of gene expression demonstrate the functional significance of TE-derived exons in presumably regulating gene expression in their respective cell types. Our data further suggests that transposable elements that donate coding sequences to human genes have been epigenetically modified in such a way as to facilitate their role in driving gene expression.

Figure 6: FPKM values for all TE-derived exons were sorted and binned in 100 equal sized bins and plotted against DHS clusters, CAGE clusters, and various histone modifications that mark promoters, TSS and exons in the GM12878 cell line.

Figure 7: FPKM values for all TE-derived exons were sorted and binned in 100 equal sized bins and plotted against DHS clusters, CAGE clusters, and various histone modifications that mark promoters, TSS and exons in the K562 cell line.
Our data suggests that TEs have a substantial contribution in donating coding sequences to human genes. The level of contribution is made apparent by transcriptome assembly without the aid of a reference transcriptome and far exceeds that provided by the reference transcriptome. TE-derived exons and transcripts are also epigenetically regulated in such a way as to potentially facilitate the establishment of cell type specific gene expression. Our results in no way discounts or diminishes the presumed importance of TEs in non-coding regions but we make an association between exonization of TEs and epigenetic regulation of transcription. Furthermore, our interpretation of the results mainly centered on the findings from the reference guided assembly of the transcripts sequence reads as we could leverage the gene model as annotation for associating with the epigenetic data. Thus, one can ascertain that there are potentially novel transcripts from the non-reference guided assembly that are presumably as important as the epigenetically regulated ones we detected. Further investigation into these potential transcript regulators is of interest and we are also considering computational ways to discern false positive and false negative detection of TE exonization using synthetic reads and/or spike-ins in the absence of the ground truth.
We thank Yuxia Cui and Harriet Kinyamu for their critical review of the manuscript. We also thank I. King Jordan for valuable insight to this work. This research was supported, in part by, the Intramural Research Program of the National Institutes of Health (NIH) and National Institute of Environmental Health Sciences (NIEHS) [Z01 ES102345-04].
The authors declare that they have no competing interests.
AH and PRB conceived the study and designed the analysis plans. AH conducted the data analyses, PRB reviewed the results and statistical analysis, AH and PRB wrote the manuscript.